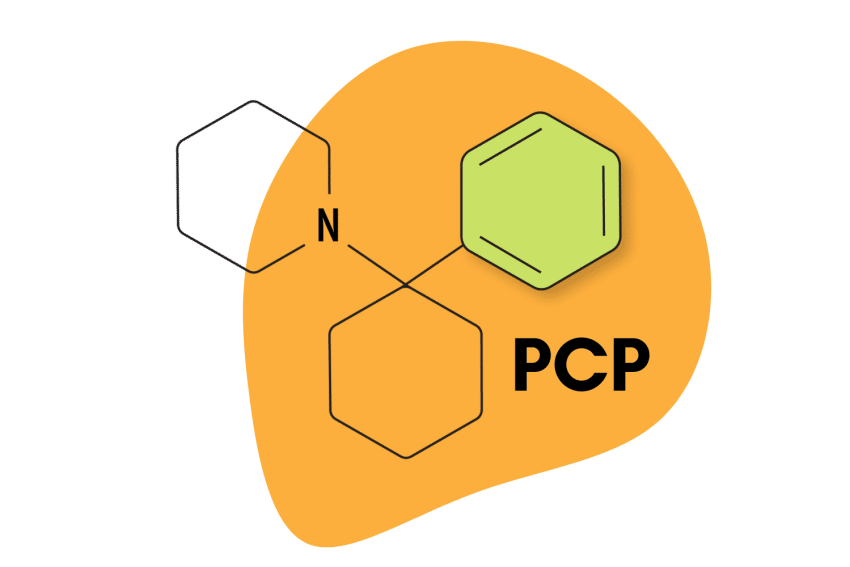
Arylcyclohexylamines
Arylcyclohexylamines are a class of dissociative drugs that cause a person to feel detached from reality. Their effects are profound and nonsensical — almost like a dream.


The most famous members of the arylcyclohexylamines include PCP (phencyclidine), ketamine, MXE (methoxetamine), DCK (deschloroketamine), and O-PCE — but there are dozens of others too.
Most of the drugs in this class are considered dissociative anesthetics — which means they produce variable psychoactive effects in lower doses and dissociate the mind from the body in higher doses. This dissociated state is extremely psychedelic and can dramatically alter the way one perceives time and space.
Some of these drugs are used as medicine; most are used purely for recreational purposes.
In this guide, we’ll explore the imaginative and trippy world of arylcyclohexylamines. We’ll explore the chemistry, safety, and subjective effects of over 35 individual arylcyclohexylamine drugs.
Arylcyclohexylamine Summary
- Chemical Structure: Contains a cyclohexyl group fused to an amine and aromatic ring
- History: Arylcyclohexylamines were originally developed by Parke Davis for use as anesthetics
- Common Examples: PCP and ketamine are the most common arylcyclohexylamines, but there are hundreds of others
- Legality: Only ketamine is approved for use in medicine. Most arylcyclohexylamines are classified as research chemicals or designer drugs
- Effect Profile: Arylcyclohexylamines are dissociative psychedelics, stimulants, and anesthetics
- Mechanism of Action: Arylcyclohexylamines primarily work by blocking NMDA receptors. They also activate mu-, delta-, and kappa-opioid receptors, sigma-1 and sigma-2 receptors, and inhibit dopamine, serotonin, and norepinephrine transporters as well as nicotinic acetylcholine receptors (nACh) receptors
What Are Arylcyclohexylamine Psychedelics?
Arylcyclohexylamines (AKA arylcyclohexan-amines) are a class of dissociative drugs that decouple one’s first-person, conscious experience from objective reality. They isolate self-aware aspects of the mind from sensory and emotional processing. Users remain fully conscious but lose all physical and emotional “noise” produced by the unconscious mind and physical body.
In lower doses, this produces a sensation similar to alcohol in which one feels as though they’re floating or on autopilot (while paradoxically remaining in control of their body). Euphoria and trippy open and closed-eye visuals are also common.


In higher doses, dissociatives produce profoundly altered states of consciousness in which the user experiences conditions that are hard to describe using spoken language. Users often describe watching themselves without the use of a mirror, falling into an endless abyss, a (non-threatening) confrontation with death, or a merging with the universe.
These drugs work by antagonizing (inhibiting) a receptor called NMDAR (N-methyl-D-aspartate receptor) in the brain. NMDA receptors act as critical regulators of brain activities ranging from memory formation and neuroplasticity to motor function and pain perception. This is the primary mechanism of action for the dissociative action. Most of these drugs also interact with dopamine, serotonin, norepinephrine, and acetylcholine — which leads to further changes to cognition and conscious experience.
The effects of these drugs are a bit of a paradox — they’re both stimulating and sedative; they’re neuroprotective and neurotoxic, and they’re addictive and non-addictive. It all comes down to the dose.
The arylcyclohexylamine class is large and diverse, with most of them having only been invented in the past 10–20 years. The first compound to be approved was PCP (phencyclidine) in the 1950s, but it was replaced ten years later by ketamine which is considered safer and more reliable.
The new wave of arylcyclohexylamine development arose as a result of a sudden surge in public interest for “legal” PCP and ketamine alternatives and designer drugs. Clandestine manufacturers would modify the base structure of PCP and ketamine to produce new psychedelics. The motivation for this spurge in development was to avoid prohibition.
Arylcyclohexylamine Chemistry
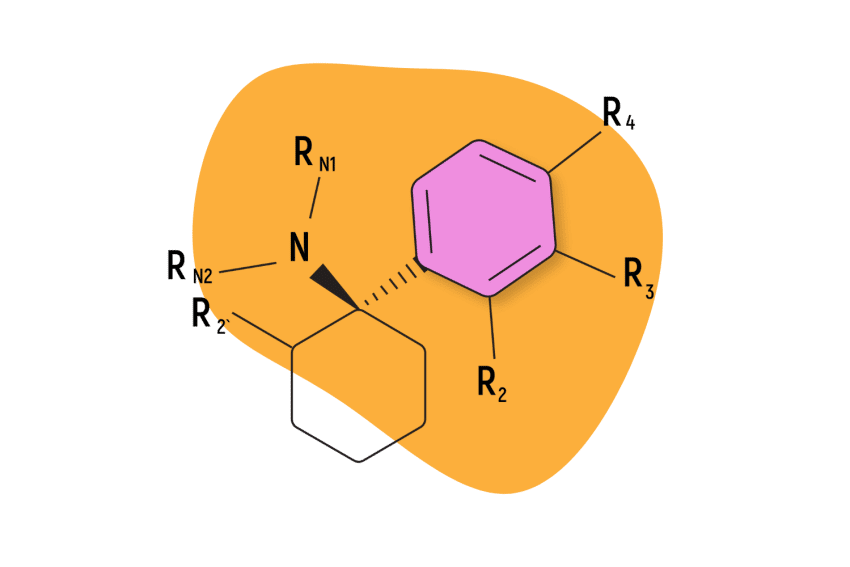
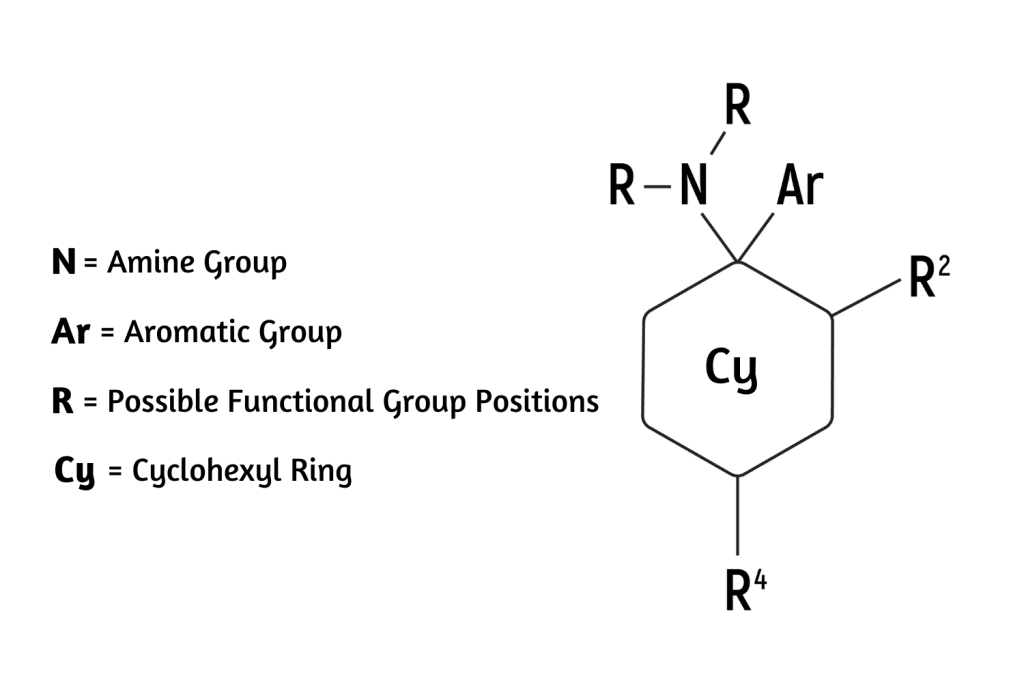
The arylcyclohexylamine scaffold contains three regions: an aromatic ring, a cyclohexane ring substitution, and an amine group.
Let’s break this down a bit.
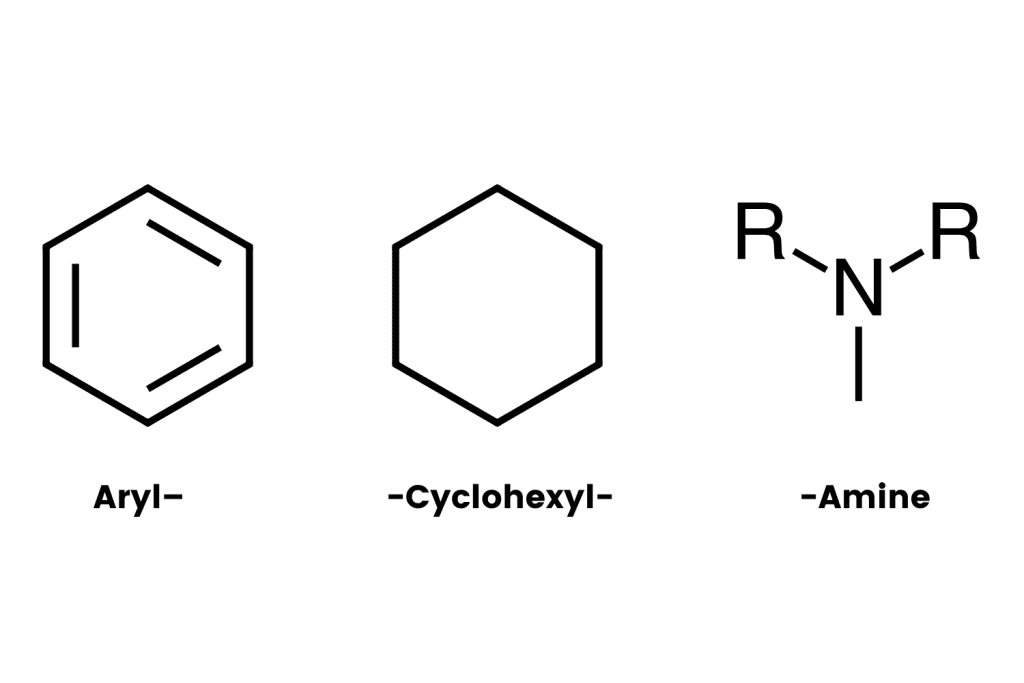
The “aryl-” refers to the presence of an aromatic ring (such as benzene or phenyl). This is one of the most stable chemical structures possible. It’s a feature found in many psychoactive molecules, including benzodiazepines, 2CX compounds, and indole alkaloids like psilocybin and DMT (dimethyltryptamine).
The “-cyclohexyl-” implies the presence of a cyclohexane molecule — a separate six-carbon ring structure forming a hexagon. This group is needed to classify a compound as an arylcyclohexylamine and it appears this group is necessary for the distinct dissociative effect. Whenever this group is removed, researchers note a substantial drop in activity at the NMDA receptors.
The final segment, “-amine” refers to the inclusion of an organic nitrogen-containing molecule.
All arylcyclohexylamines maintain this same base formula, but alterations can be made by attaching different molecules (called moieties) to the aromatic ring, the amine group, or the cyclohexane ring at position 2.
There are hundreds of possibilities for new arylcyclohexylamine molecules, but not all of them are psychoactive, and some are directly toxic.
Below, we explore the compounds in this family with known psychoactive effects.
Arylcyclohexylamine Naming Conventions
There’s a general formula used to denote the name of a given arylcyclohexylamine — X-YCZ:
- X — refers to the substitutions made to the molecule and the site it’s attached to.
- Y — refers to the aromatic ring. In most cases, this is a P for “phenyl,” but there are other compounds that can be used here, such as thiophene.
- C — refers to the cyclohexane ring, which is always present in a true arylcyclohexylamine.
- Z — refers to the amine group, for which there are many different possibilities. This is normally shortened in some way in the final naming convention of the drug. For example, PCP contains piperidine, while PCE contains ethylamine.
Are Arylcyclohexylamines Safe?
All members of the arylcyclohexylamine family carry a risk of toxic side effects and potential for abuse — some more than others.

The arylcyclohexylamine group contains several known toxic compounds, and all of them carry a moderate to high risk of abuse. High doses of arylcyclohexylamines can result in seizures, hyperthermia, and an increased risk of accidental injury due to disinhibition.
Long-term use can lead to the development of psychiatric disorders, cognitive decline (problems with memory and focus), or injury to the bladder, kidney, liver, and lungs.
There’s very little official research on the safety of individual arylcyclohexylamines. The exceptions are PCP and ketamine, which have been used as medicine since 1957 and 1970, respectively. This means the safety profile of most compounds in this class is not well defined.
Arylcyclohexylamine Overdose
Fatal overdoses on arylcyclohexylamines alone are rare. Side effects of overdose include schizophrenia-like delusions, mania, dyskinetic and catatonic movements, hypertension, and coma [22]. Overdose cases are generally well managed in a hospital, and most patients recover within 12–24 hours.
Deaths have occurred as a result of hyperthermia (elevated body temperature), acute renal failure, rhabdomyolysis (muscle cell death), and excessive blood clotting.

Without more information on the toxicity of individual arylcyclohexylamines, it’s hard to pinpoint which compounds pose the greatest risk. However, differences in potency between various compounds contribute directly to the risk associated with overdoses.
More potent compounds like PCP, PCE, 3-HO-PCE, 3′,4′-MD-PCPr, 3′-HO-PCP, 3′-MeO-PCE, 3′,4′-MD-PCE, and others carry a much greater risk than weaker and more “forgiving” compounds like ketamine, DCK, or MXE.
Bladder Damage
All arylcyclohexylamines carry a risk of urinary toxicity. Various metabolites of the arylcyclohexylamines pass through the kidneys and into the urine. Several of these metabolites, including norketamine, dehydronorketamine, hydroxyketamine, and hydroxynorketamine (HNK), are believed to be toxic to both the kidneys and the bladder epithelial cells. With repeated exposure, these metabolites lead to damage and destruction of the kidneys and the inside layer of the bladder.

This usually first appears in the form of blood in the urine, bladder pain, and pain upon urination. As the bladder walls become damaged, scar tissue forms which can cause chronic pain, reduced bladder capacity, and incontinence (inability to control urination).
This damage is most common in regular users of low-potency arylcyclohexylamines (like ketamine), but it’s also been reported in users after just a few sessions. Higher potency analogs have a lower chance of causing bladder damage simply because users need to take less to achieve the same effects. Less substance means less toxic metabolites end up in the bladder.
With that said, bladder injury has been reported with just about every arylcyclohexylamine discussed in this guide.
Brain Damage
One of the biggest worries about the repeated use of NMDA antagonists (including, but not limited to, the arylcyclohexylamines) is the formation of Olney’s lesions in the brain. Olney’s lesions are characterized by “pockets” of damaged white matter — resulting in cognitive deficits like memory loss, reduced capacity for learning, and difficulty concentrating or staying focused.

Loss of white matter in the brain is also a key factor in the progression of Alzheimer’s disease and Huntington’s disease.
This side effect is complicated. Low-dose ketamine and other NMDA antagonists have been shown to have the opposite effect — they act as neuroprotectants [24], which protect the brain from brain damage. Ketamine is sometimes used by doctors to protect the brain after a stroke and may even help recover damaged neurons [25].
The key differentiator here is the dose and frequency of use. It appears that moderation is the best defense against brain damage induced by these chemicals. Lower doses and plenty of time off between doses do not appear to cause Olney’s lesions.
Arylcyclohexylamine Addiction & Abuse
While it’s entirely possible to use arylcyclohexylamines responsibly in moderation, all members of this group carry a moderate risk of abuse and physical dependence.
Arylcyclohexylamines are very powerful. They’re some of the strongest psychedelics currently known. They do this without the anxiety or fear reaction that comes along with most psychedelic drugs. Their ability to disconnect the ‘conscious self’ from objective reality allows users to reframe their perception of what it means to be conscious beings. It also temporarily removes the pain attached to one’s emotional being. Users enter a state of “euphoric nothingness.”
All the shame, guilt, and pain accumulated throughout life are (temporarily) lifted off one’s shoulders. This feeling can become addictive — especially in people who have experienced a lot of trauma and turmoil in their life.
Dissociative abuse doesn’t happen overnight. Most users start dabbling with dissociatives responsibly and often with the right intention, but it silently evolves into abuse over several weeks, months, or years.

Addiction is defined as a compulsive desire to use a substance despite clear negative impacts of that drug on the body, one’s finances, or one’s relationships.
Unfortunately, part of the problem with dissociative addiction is the development of delusional thought patterns that can block people from being able to recognize or accept their own problems with addiction.
With repeated use, these drugs tend to replace one’s internal dialog with the nonsense that only the individual can understand — which only serves to further distance users from their family and friends and gives them even more reason to seek the temporary escape these drugs can provide.
The Inherent Risks of Research Chemicals
By definition, research chemicals have gone through little to no formal testing to prove they’re safe for human consumption. This lack of testing carries a variety of risks.
Drug companies producing new medicines have to jump through a series of hoops before the drug is officially approved. These hoops are designed to, first and foremost, prove that the medicine is nontoxic in the recommended dosages.
Secondly, the drug needs to be proven effective for the target condition.
This requires many years of focused research, specialty equipment run by professional researchers, and potentially hundreds of millions of dollars. The average cost of drug development in the United States is estimated to be around $350 million — but it’s not uncommon for research costs to exceed $1 Billion.
PCP and ketamine are the only members of the arylcyclohexylamine family to have undergone this formal testing. Everything else here is classified as a research chemical.
Some drugs have undergone preliminary testing by independent researchers, and many of them have anecdotal testing available in the form of trip reports. Needless to say, none of this is enough to prove these drugs don’t carry significant risks. Most arylcyclohexylamine research chemicals have only been on the market since 2008, so there simply hasn’t been enough time to properly assess the long-term health consequences of these drugs.

Anybody taking a research chemical should be aware of these inherent risks. They should know that there’s virtually no guarantee of safety when taking these molecules — especially if they happen to be sensitive to the drug or are taking other medications or substances alongside it. Side effects are common and unpredictable, and the long-term impact of these drugs on one’s mental and physical health remains a black box.
Other risks are the formation of unknown (potentially toxic) metabolic byproducts as the drug is processed by the body (such as with 3-F-PCP or other fluorinated compounds). Without adequate research, it’s impossible to know exactly what happens to these drugs after they enter the body.
Other factors affecting the safety of these drugs are the abundance of adulterated or contaminated products on the designer drug market, as well as improper labeling, and the use of unclean needles used for injection.
Are Arylcyclohexylamines Legal?
Yes and no. Arylcyclohexylamine analogs of PCP are classified as a Schedule I restricted substances in the United States. Most other countries consider PCP and its analogs illegal under equivalent scheduling laws.
This may not apply to ketamine analogs. This is because the Federal Analog Act — which makes any non-medically-approved analog of a Schedule I or Schedule II drug automatically illegal — likely doesn’t apply to ketamine analogs. Ketamine is a Schedule III drug, which excludes it from the details of the analog act.

This only applies to possession — the unregulated sale of ketamine or ketamine analogs remains highly illegal in the United States and around the world.
Despite introducing the analog act nearly 40 years ago, vendors selling arylcyclohexylamines are thriving online. The argument is that only specific compounds are included on the list of restricted substances. By modifying the chemical structure slightly, vendors believe selling these drugs is legal through loopholes in federal law. This has been a driving force in the rapid development of new research chemicals in the arylcyclohexylamine family.
Forms of Administration
Arylcyclohexylamines can be used in virtually all forms of administration. They can be taken orally in the form of a pill or capsule, snorted, injected into the muscles, or directly into the veins, smoked, or vaporized.
Each method comes with its own set of pros and cons. Oral tablets are the easiest and safest to consume, but they take longer to kick in and aren’t as bioavailable as injections. Likewise, injections kick in quickly but can be dangerous to administer without sterile medical equipment and an understanding of the dose and how to inject the drug safely.
Bioavailability of various arylcyclohexylamine dosage forms:
| Route | Bioavailability (%) |
| Oral | 17–24% |
| Sublingual | 24–30% |
| Intranasal | 25–50% |
| Intramuscular (IM) | 93% |
| Intravenous (IV) | 100% |
1. Oral
Taking ketamine orally means swallowing a pill or capsule. Some users will wrap the powder in a rolling paper if they don’t have a capsule — this practice is referred to as parachuting. Others mix the powder into a drink.

The downside of oral use is that this form offers the lowest bioavailability (refers to the amount of a drug that’s absorbed into the body after it’s consumed). This means the dosages for oral consumption are going to be the highest overall — usually around 10–20% higher than the dose for insufflation (snorting).
For ketamine and other lipid-soluble arylcyclohexylamines, the onset time for oral consumption is around 20 or 30 minutes.
2. Sublingual
Sublingual administration involves the absorption of drugs through the microcapillaries lining the underside of the tongue and mouth. Here, tiny blood vessels located near the surface allow the passage of various drugs directly into the bloodstream.
This is how drugs like LSD are normally administered. By holding the LSD tab under the tongue for several minutes, the active ingredients transfer into the blood vessels — offering a relatively fast onset of effects and bypassing first-pass metabolism. Drugs taken orally are sent to the liver, which attempts to neutralize as much of the active ingredients as possible before entering circulation. By avoiding first-pass metabolism via the liver, the potency of a given drug is preserved.

Sublingual ketamine is common for low-dose at-home treatments in the form of a lozenge containing between 25 and 75 mg of ketamine each — taken every couple of days in between integration sessions.
For ketamine (and likely other arylcyclohexylamines), the onset time for sublingual administration is around 10–15 minutes.
3. Insufflation (Snorting)
Most recreational users snort arylcyclohexylamines because it offers the best blend of relative safety (no injections) and dose efficiency (the effects kick in stronger and hit harder than oral consumption).
The problem with insufflation is that the crystals can cause serious damage to the nasal cavity, especially with repeated use. Some compounds are worse than others, and it depends on how finely ground the powder was before ingestion.
Drugs that require larger doses, such as 3-MeO-PCMo or 2-BrDCK, pose the biggest risk to the nasal cavity and tend to cause a lot of pain. Drugs with a much lower dose require less powder to reach the desired effects and are generally preferred for intranasal use.

Johnson & Johnson created a nasal ketamine spray called Spravato® (esktamine) that has been well-received by the medical community as a means of administering ketamine to children and adults alike. The spray doesn’t burn the nose and is rapidly absorbed within a couple of minutes.
Most arylcyclohexylamines kick in within 5 or 10 minutes after snorting them.
4. Intramuscular Injection (IM)
People who take these compounds intending to reach a K-hole state most often turn to intramuscular or intravenous injections. These modes of administration offer the fastest onset of effects and ensure very high bioavailability.

IM injections carry a substantially higher level of risk than snorting or taking orally. Contaminated needles can lead to infections and mistakes with dosages can lead to experiences that are significantly more intense than expected. Source material contaminated with other drugs, such as fentanyl, benzodiazepines, amphetamines, or other compounds, can lead to serious side effects in a very short period of time.
Most arylcyclohexylamines kick in within 2 or 3 minutes when used intramuscularly.
5. Intravenous Injection
IV injection is the most common method used to administer ketamine in hospitals. The dose can be precisely measured and titrated to ensure consistent anesthesia for as long as necessary.

This form of administration carries the highest risk of introducing blood-borne pathogens like HIV or hepatitis. IV injections also carry the highest risk of resulting in overdoses if the dosage isn’t measured with precise accuracy.
Drugs administered via IV usually kick in within 1 minute or less.
6. Vaped or Smoked
Many members of the arylcyclohexylamine family can be vaped or smoked as well. In fact, one of the most common ways PCP is used today is by dipping a cigarette in a liquid solution of PCP.
Some of the PCP will be destroyed by combustion, but a lot still ends up in the lungs and subsequently transferred into the bloodstream. Despite being one of the least efficient means of administration, smoking arylcyclohexylamines is still very affordable and provides powerful effects with just a few puffs. It costs an average of $5 to dip a cigarette in PCP, which can contain anywhere from 2–30 mg each.

Vaporizing arylcyclohexylamines is another popular means of administration because users can control the temperature to prevent degradation of the active ingredients due to extreme heat. Mesh-style vaporizers work best for this because you can apply the powder directly to the mesh heat screen. This also allows users to carefully and accurately measure out the desired dose — rather than leaving this up to guesswork with dipped cigarettes.
The effects of vaping or smoking arylcyclohexylamines usually kick in within about 3 to 5 minutes.
List of Arylcyclohexylamines
| Image | Name | Status | Estimated Threshold Dose (Oral) | Estimated Duration of Effects |
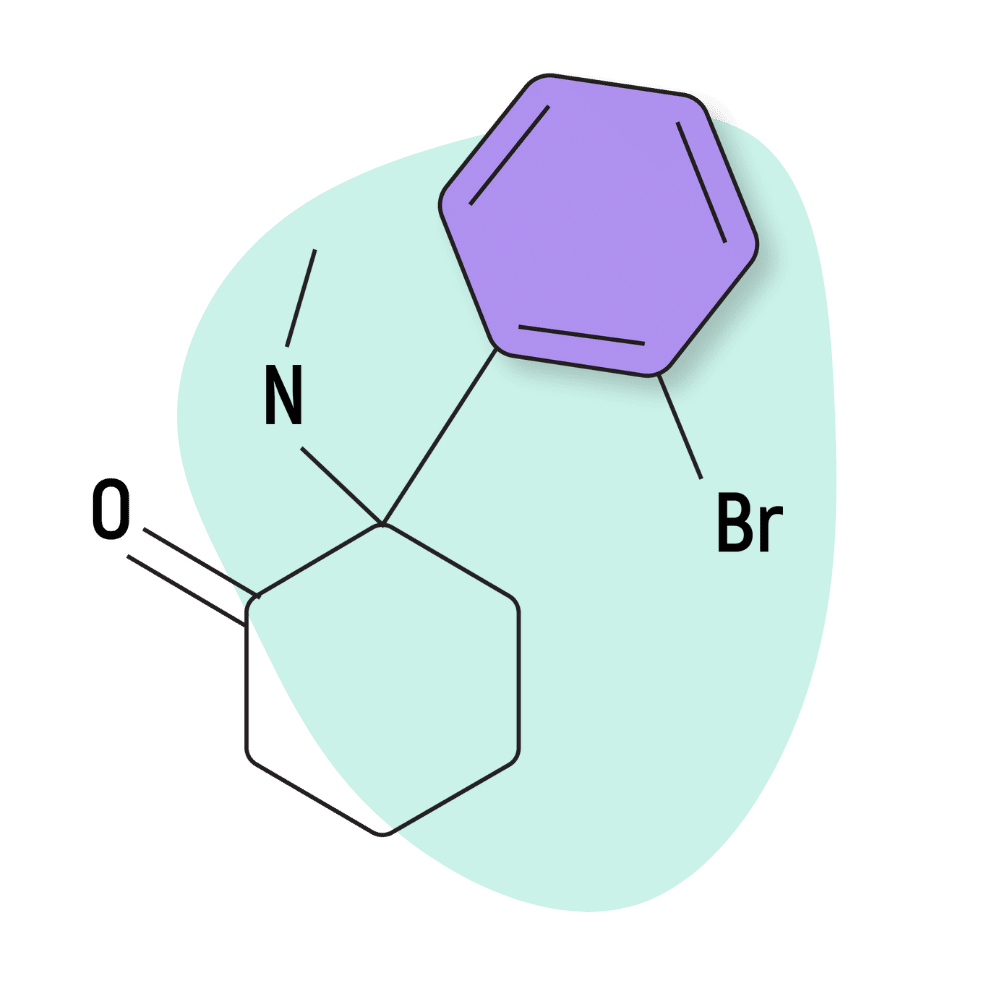 | 2-BrDCK (2-Bromodeschloroketamine) | Research Chemical 🧪 | 150 mg | 1–3 hours |
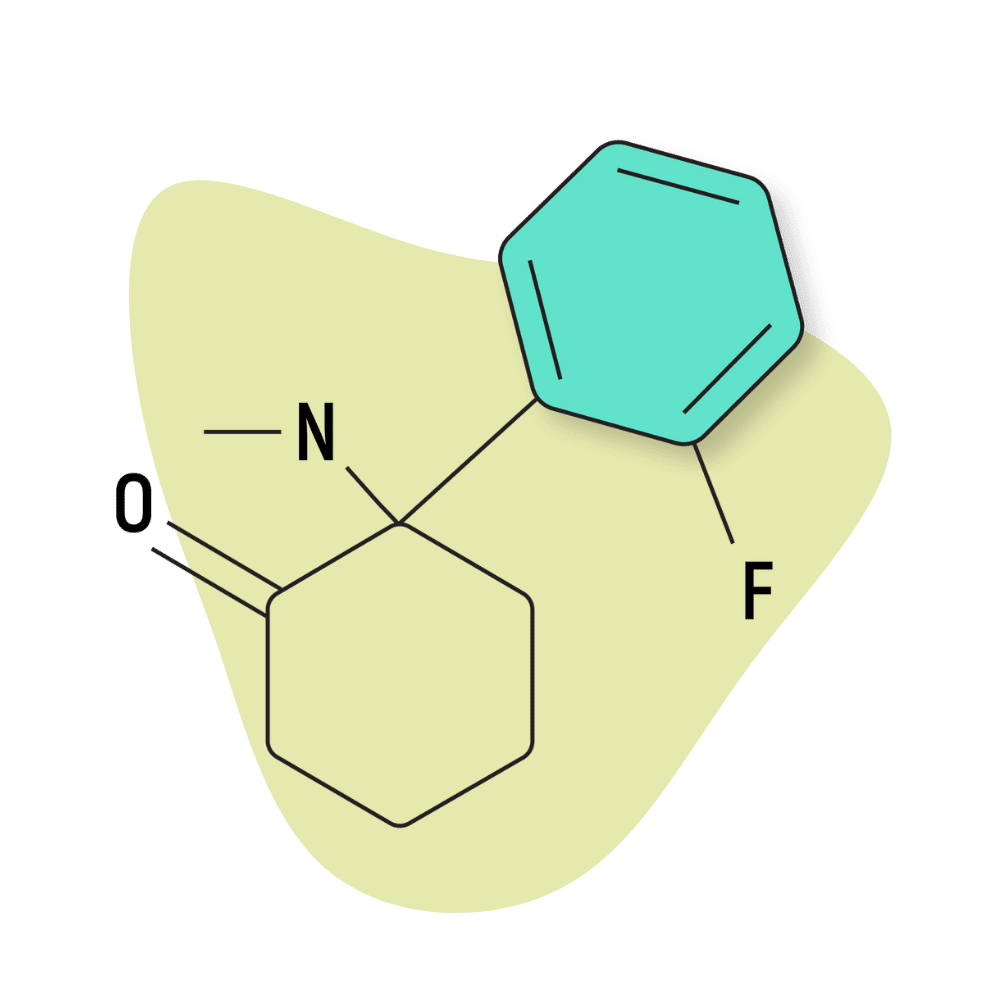 | 2-FDCK (2-Fluorodeschloroketamine) | Research Chemical 🧪 | 10 mg | 2–4 hours |
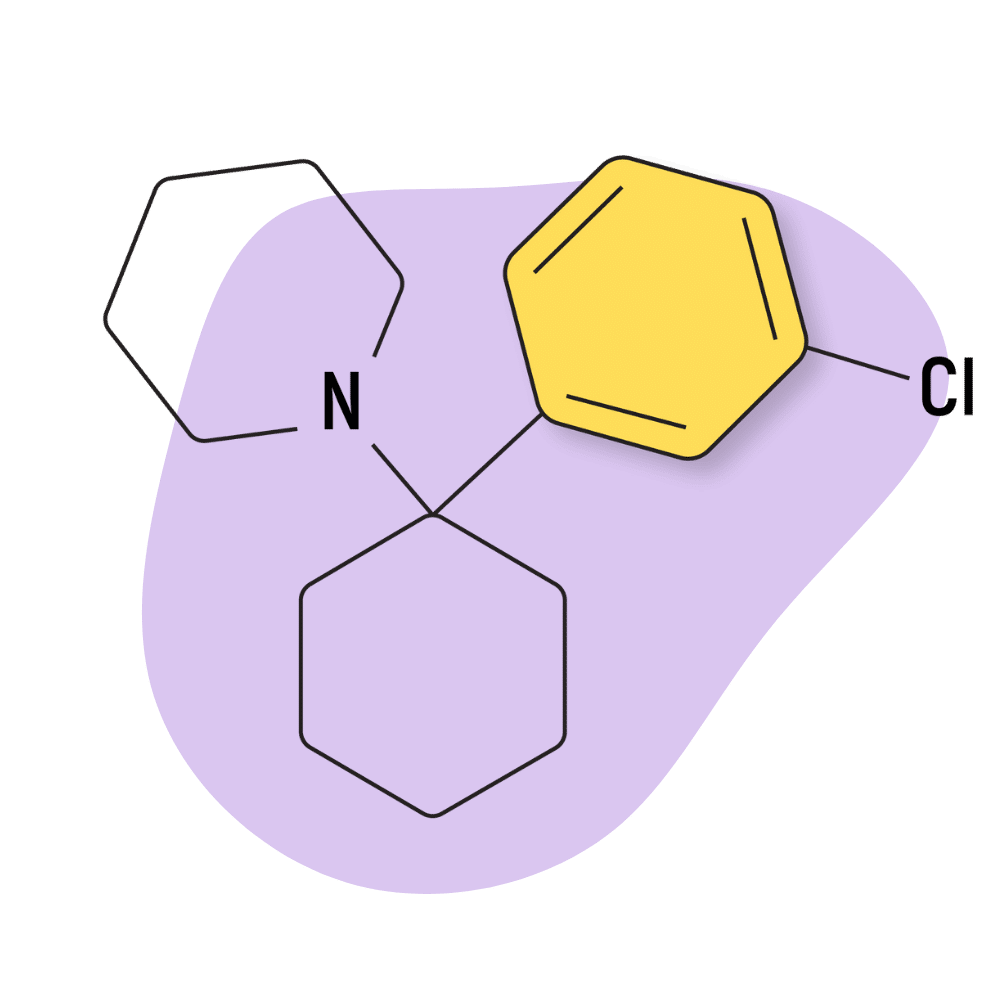 | 3′-Cl-PCP (3-Chlorophencyclidine) | Research Chemical 🧪 | 15 mg | 3–6 hours |
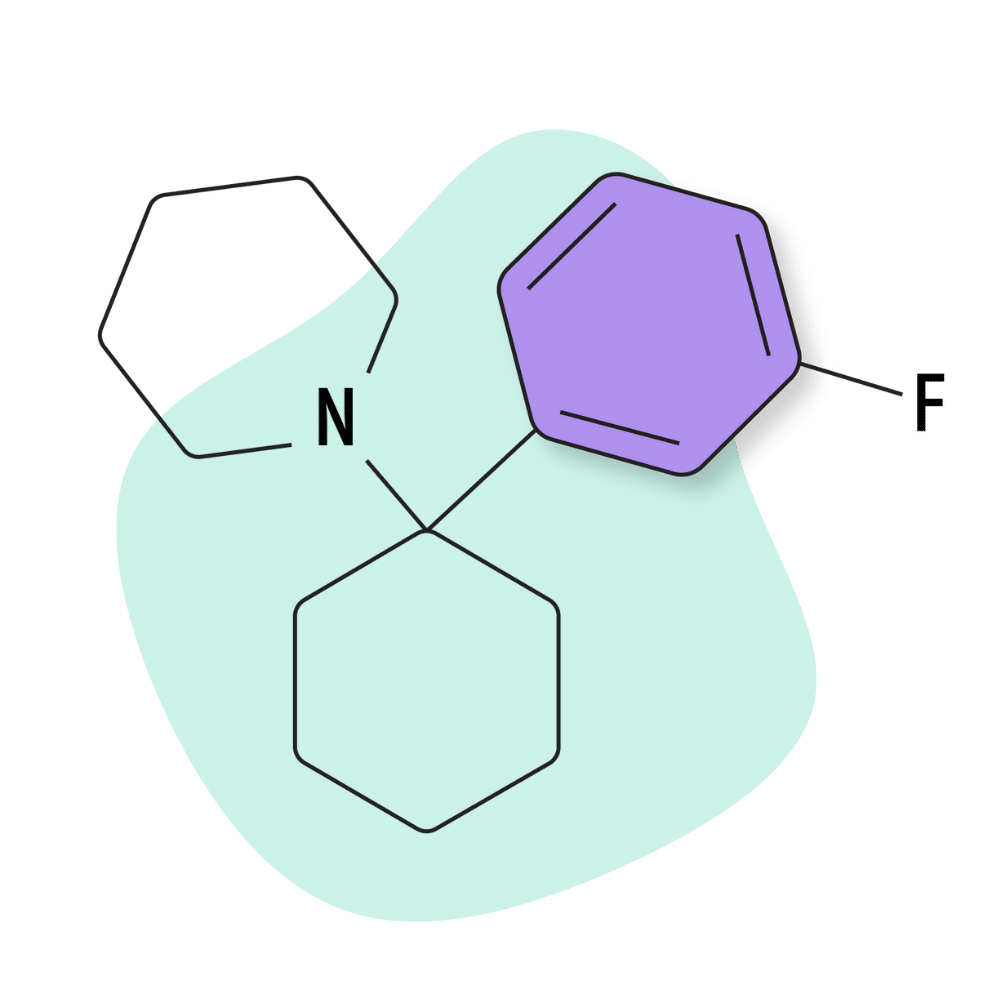 | 3′-F-PCP | Research Chemical 🧪 | 5 mg | 2–4 hours |
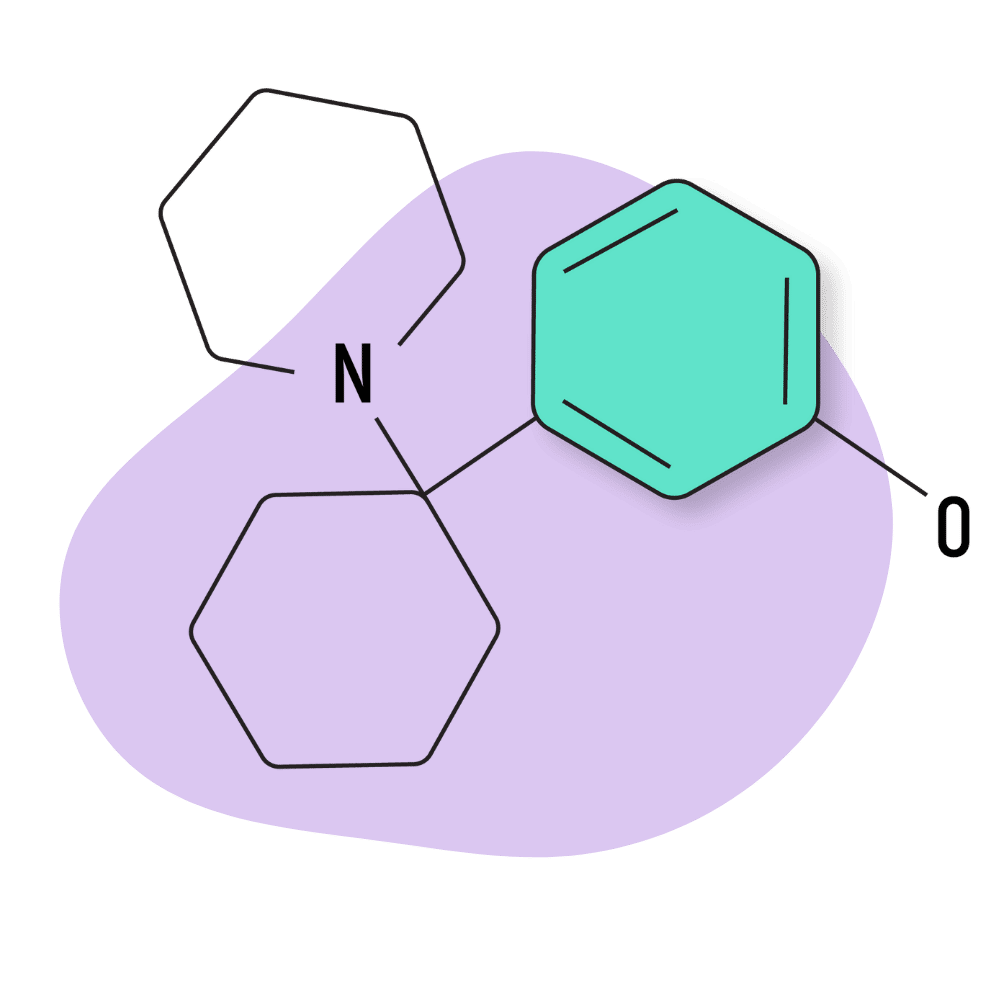 | 3′-HO-PCE | Research Chemical 🧪 | 5 mg | 3–6 hours |
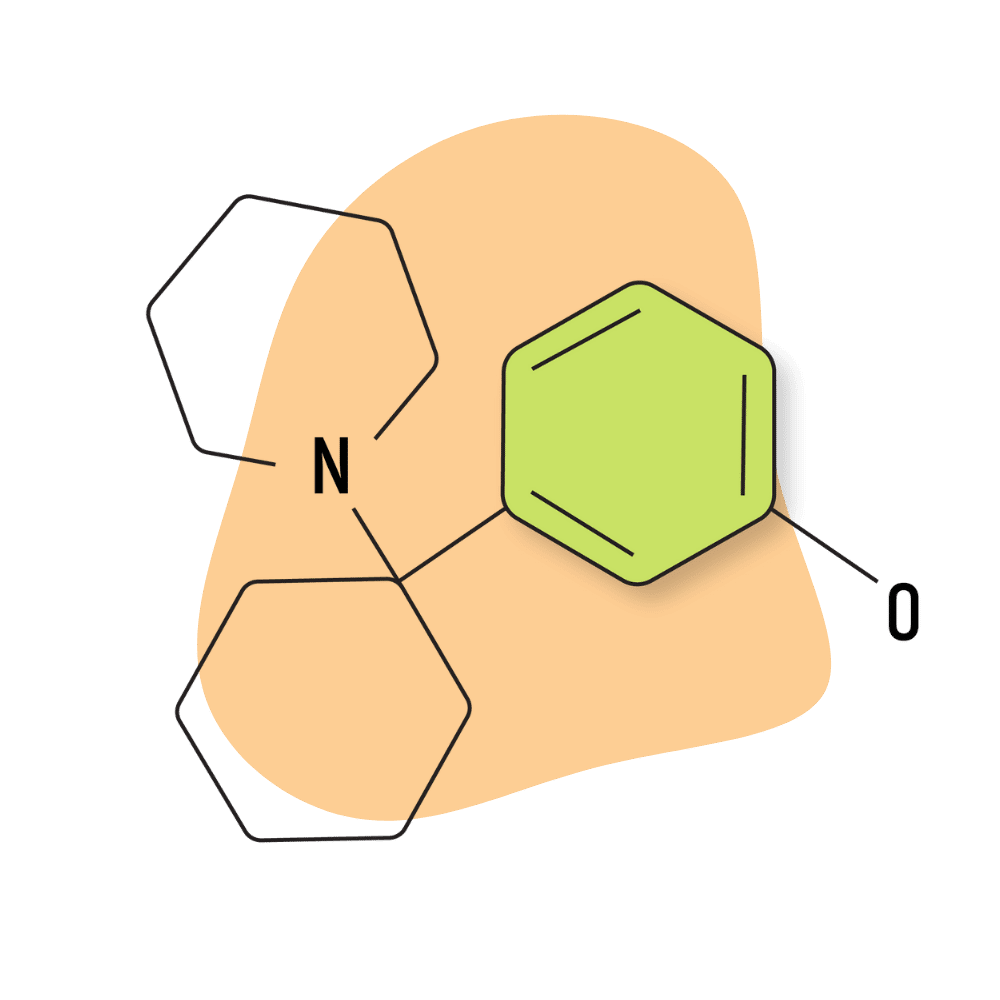 | 3′-HO-PCP | Research Chemical 🧪 | 1 mg | 3–6 hours |
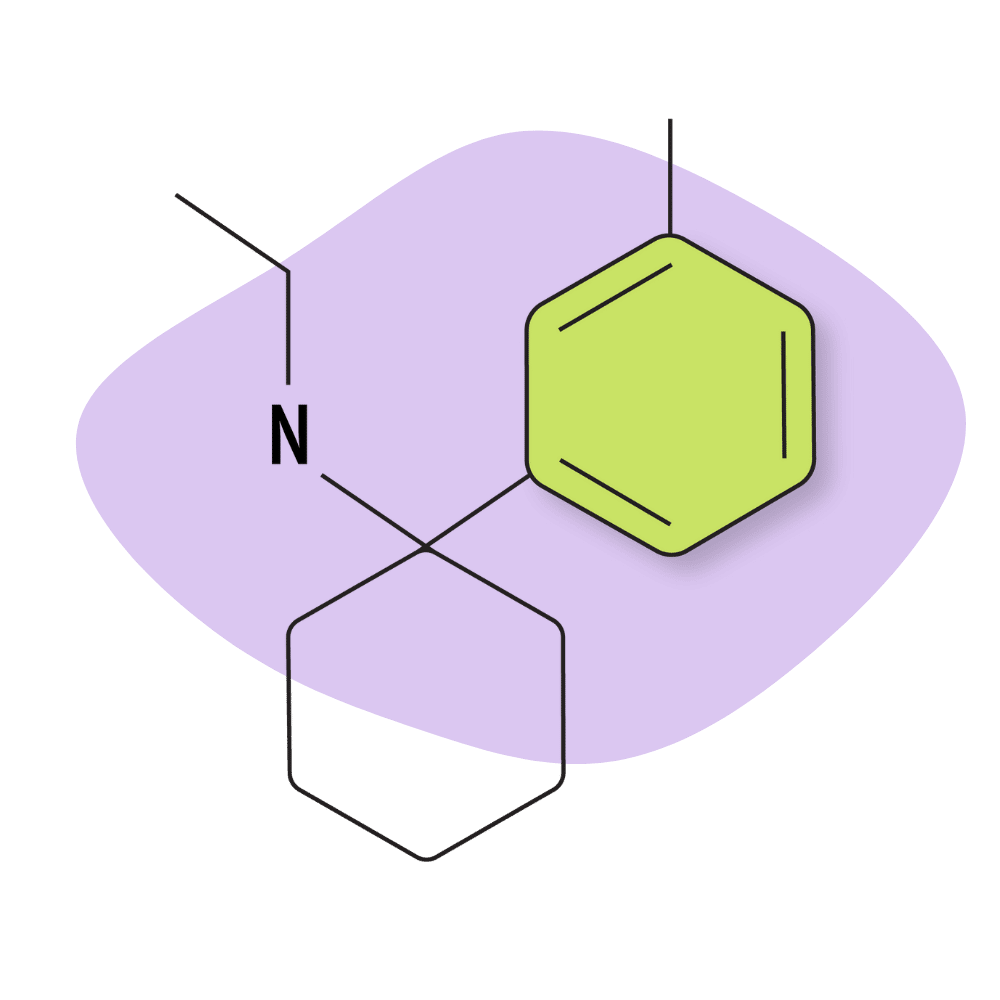 | 3′-Me-PCE | Research Chemical 🧪 | 2 mg | Unknown |
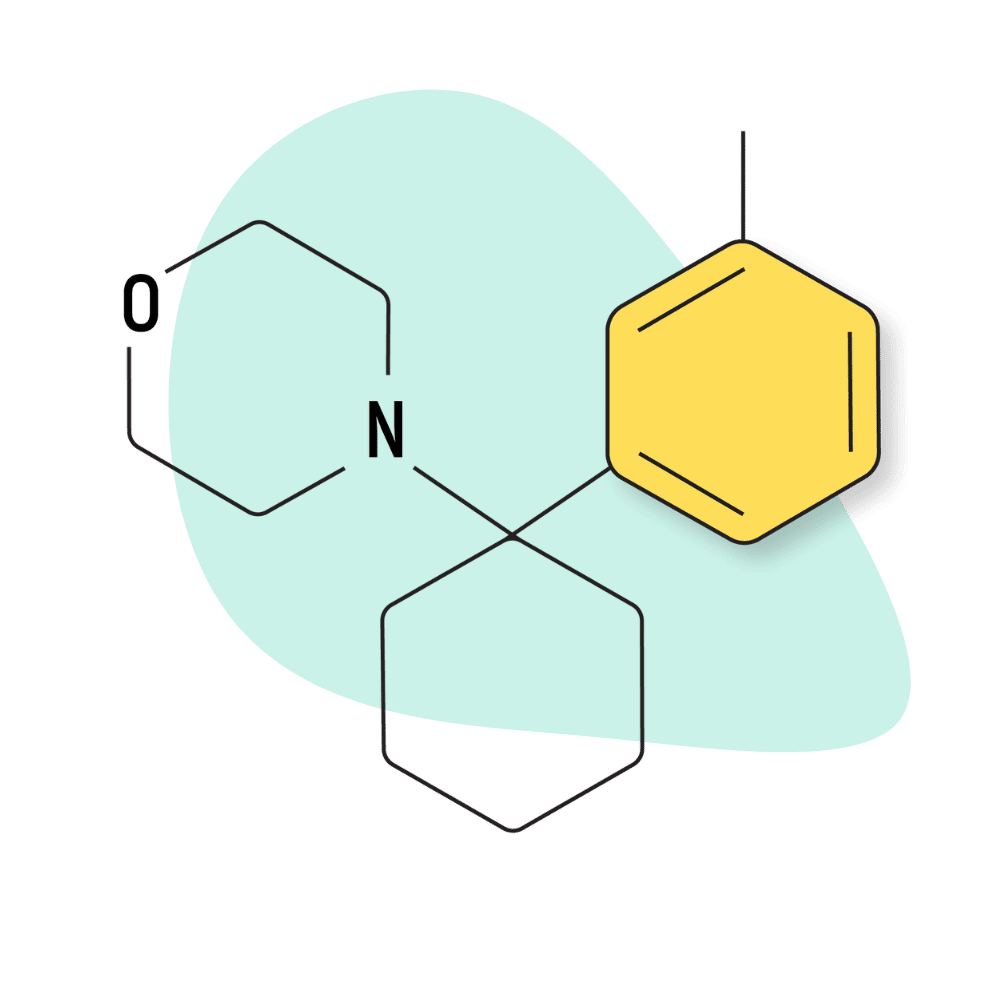 | 3′-Me-PCMo | Research Chemical 🧪 | Unknown | Unknown |
 | 3′-Me-PCP | Research Chemical 🧪 | 2 mg | Unknown |
 | 3′-Me-PCPr | Research Chemical 🧪 | 1 mg | Unknown |
 | 3′-Me-PCPy | Research Chemical 🧪 | 3 mg | 3–6 hours |
 | 3′-MeO-PCE | Research Chemical 🧪 | 3 mg | 3–5 hours |
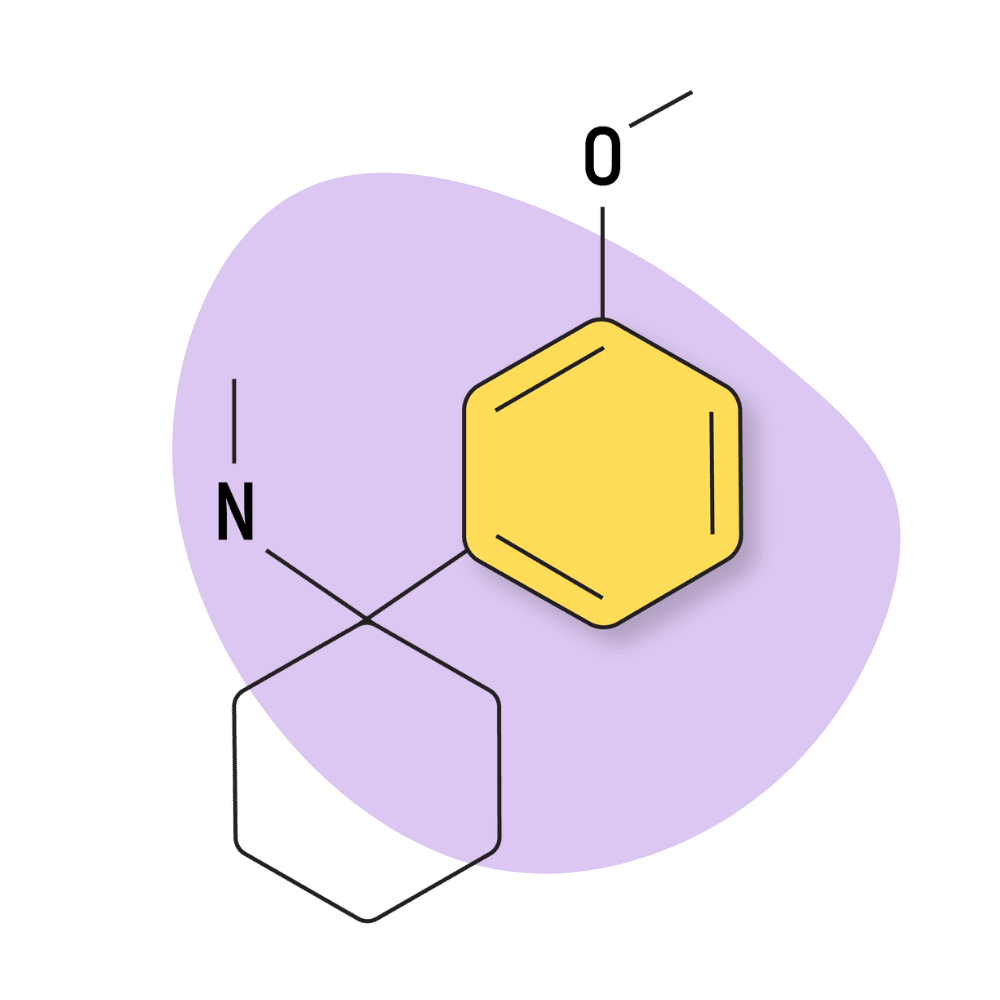 | 3′-MeO-PCMe | Research Chemical 🧪 | 8 mg | Unknown |
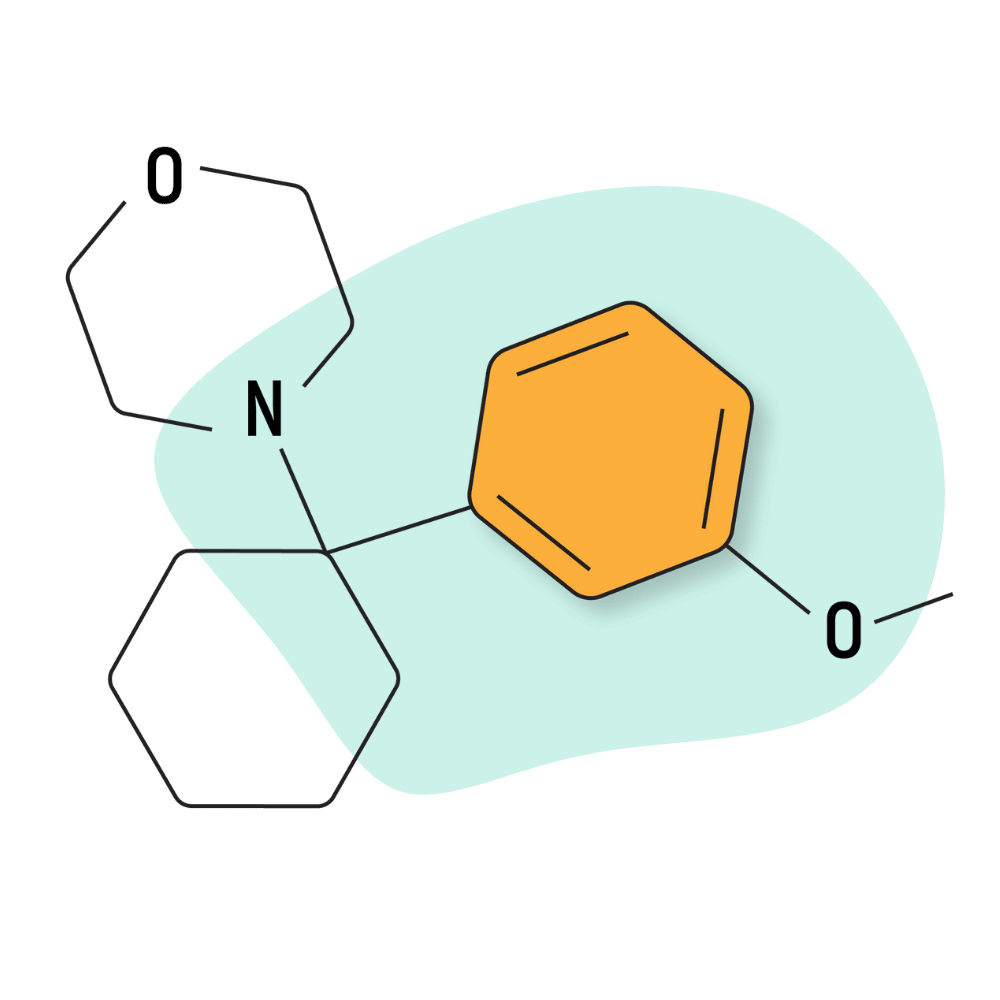 | 3′-MeO-PCMo | Research Chemical 🧪 | 50 mg | Unknown |
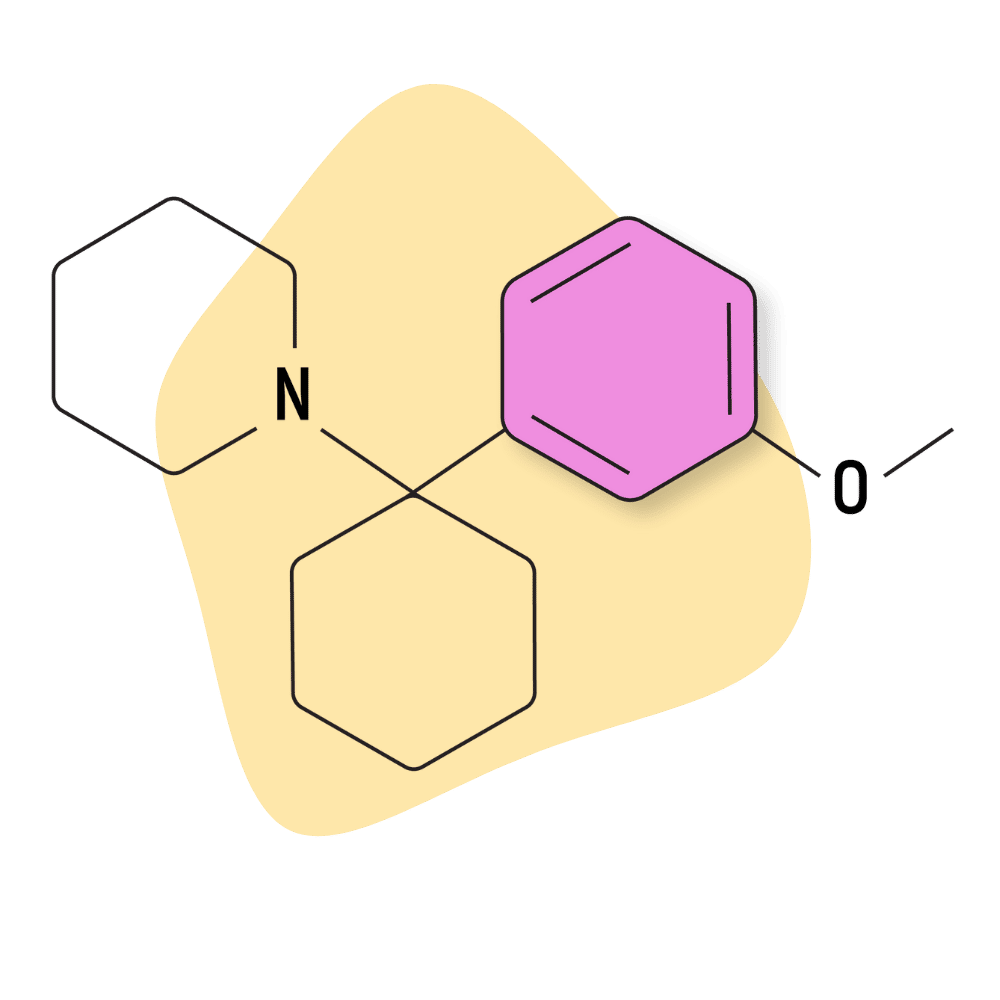 | 3′-MeO-PCP | Research Chemical 🧪 | 1 mg | 3–5 hours |
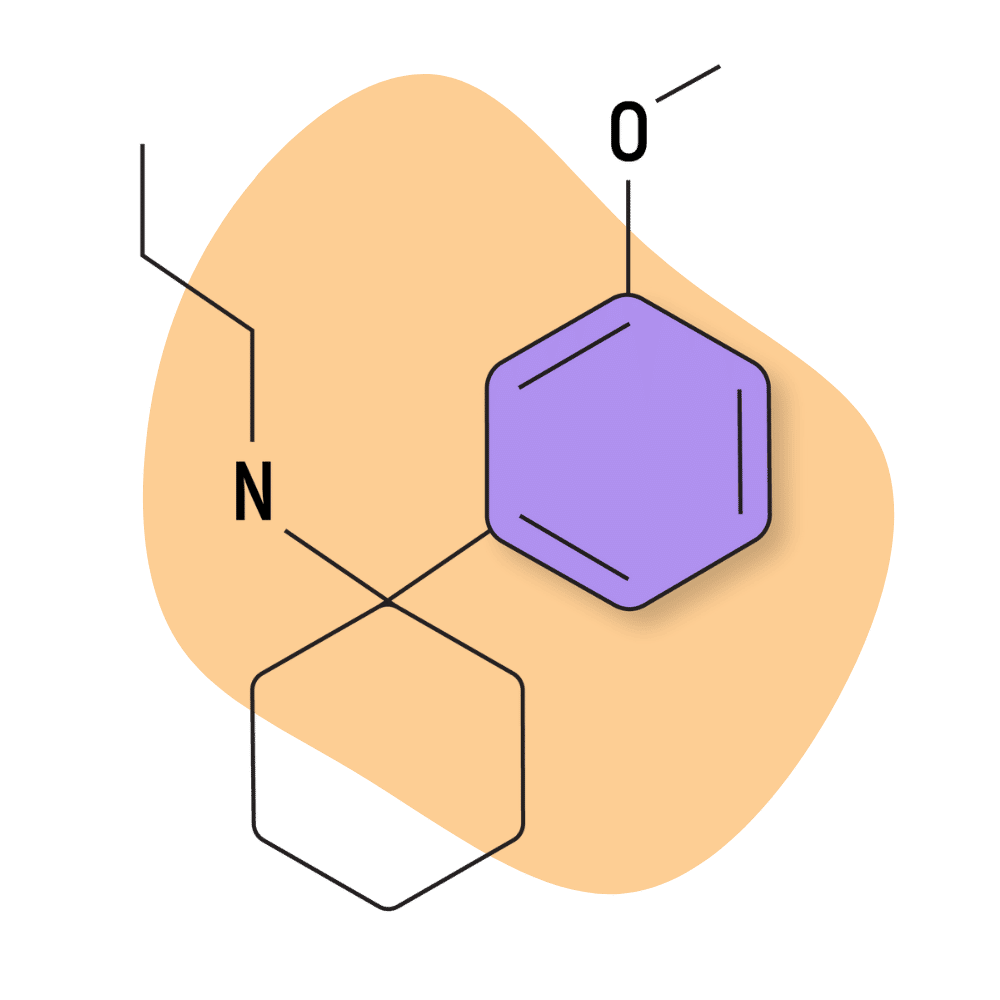 | 3′-MeO-PCPr | Research Chemical 🧪 | 2 mg | Unknown |
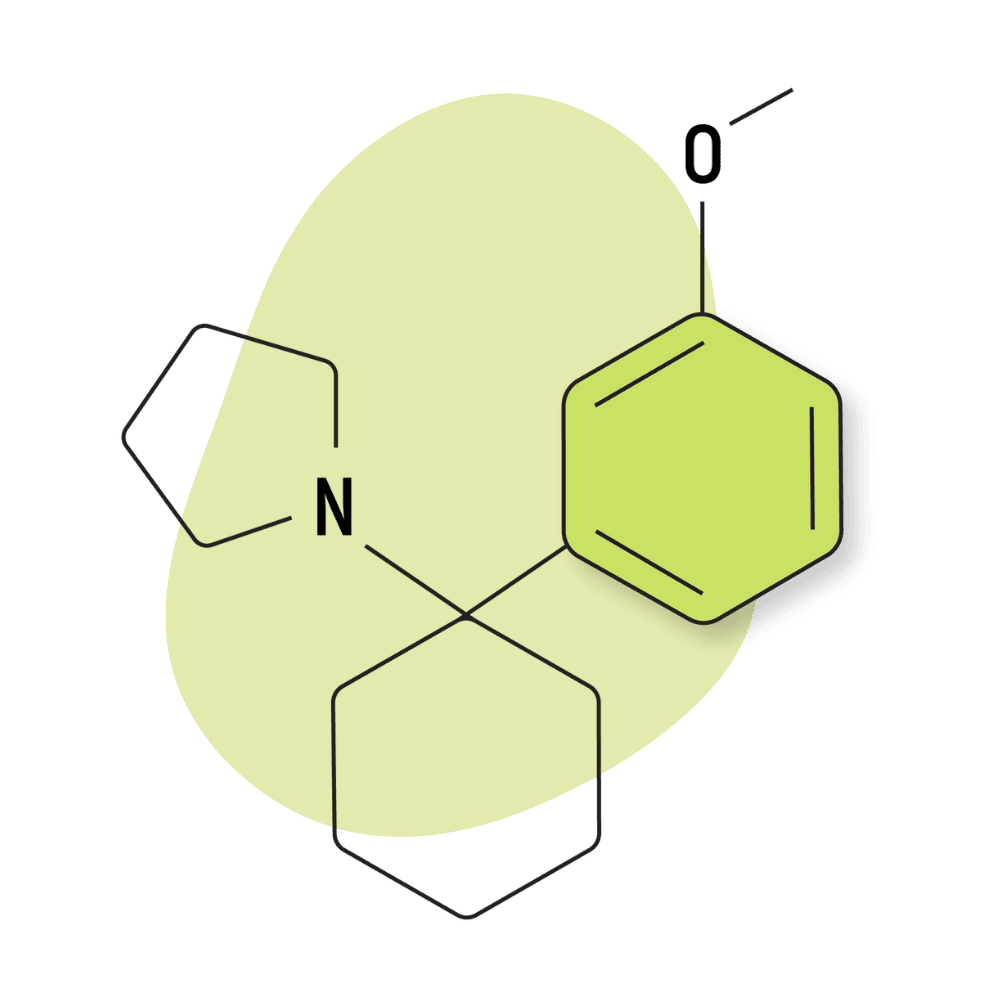 | 3′-MeO-PCPy | Research Chemical 🧪 | 2 mg | Unknown |
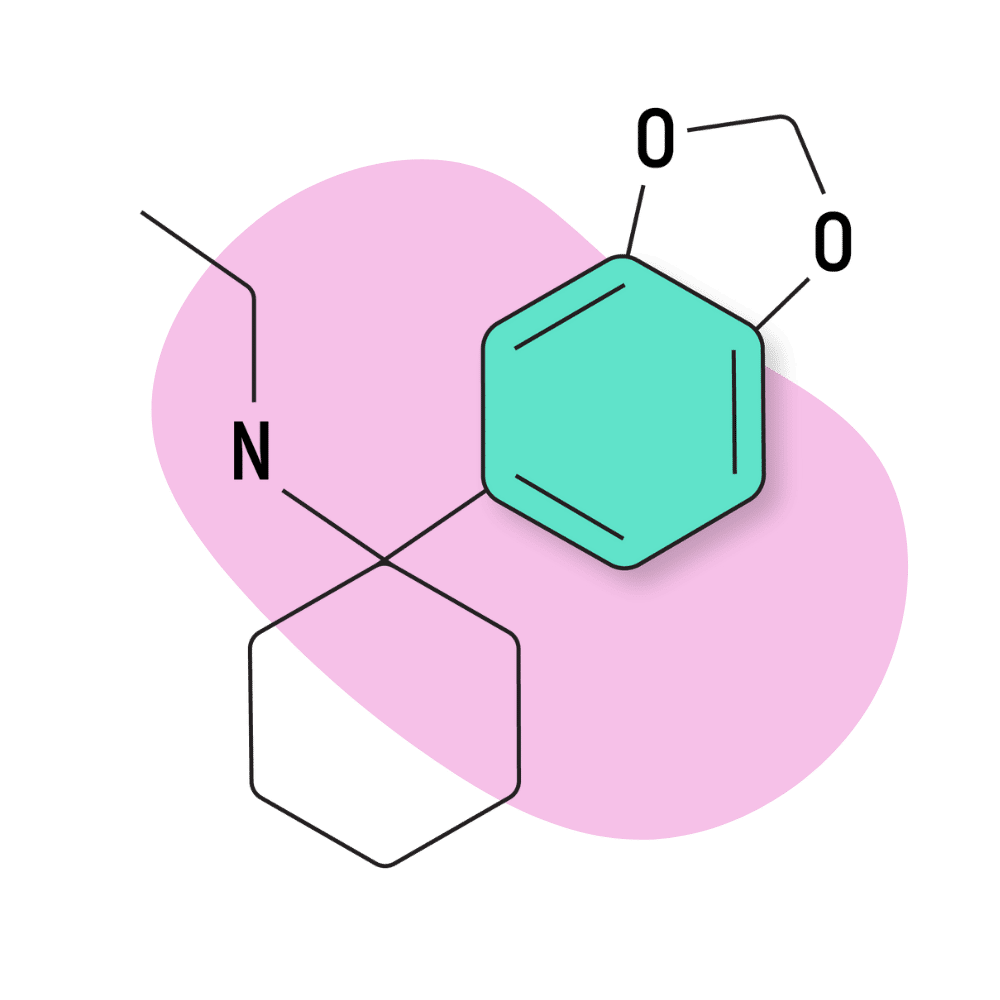 | 3′,4′-MD-PCE | Research Chemical 🧪 | 2 mg | 3–5 hours |
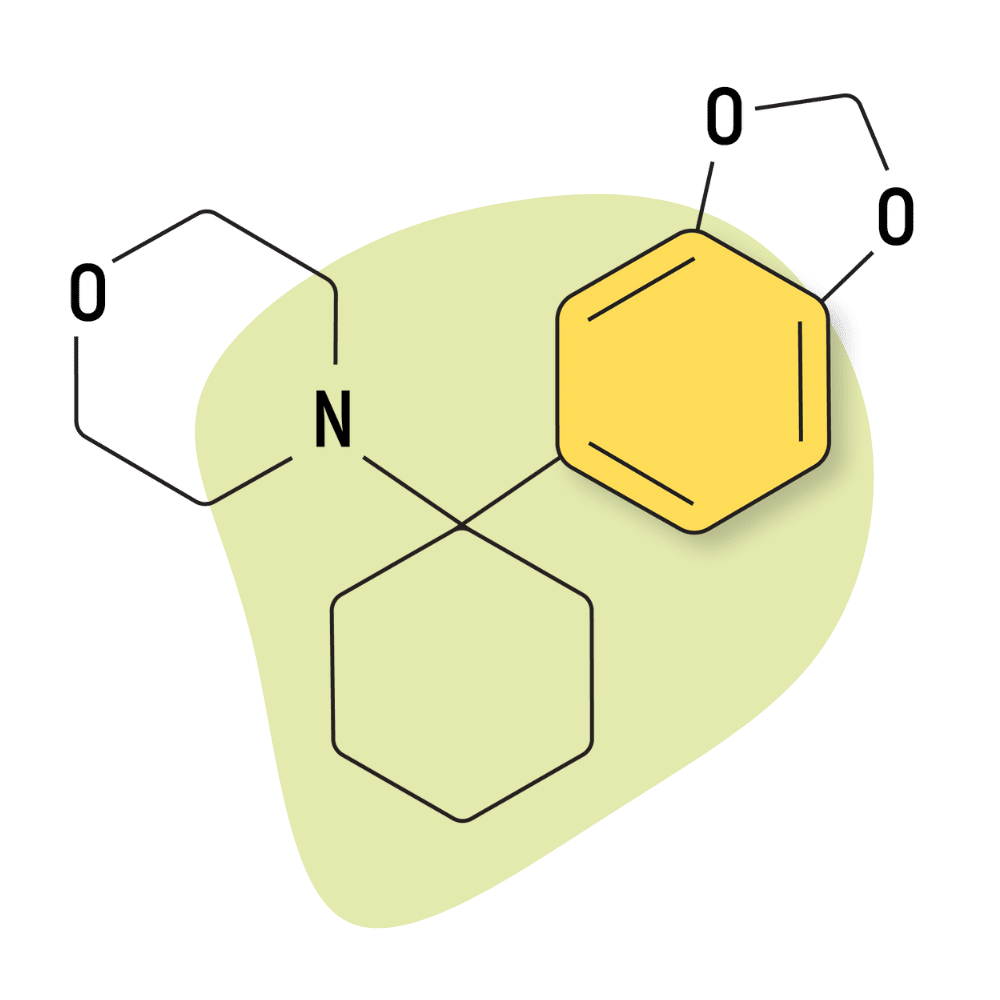 | 3′,4′-MD-PCMo | Research Chemical 🧪 | 4 mg | Unknown |
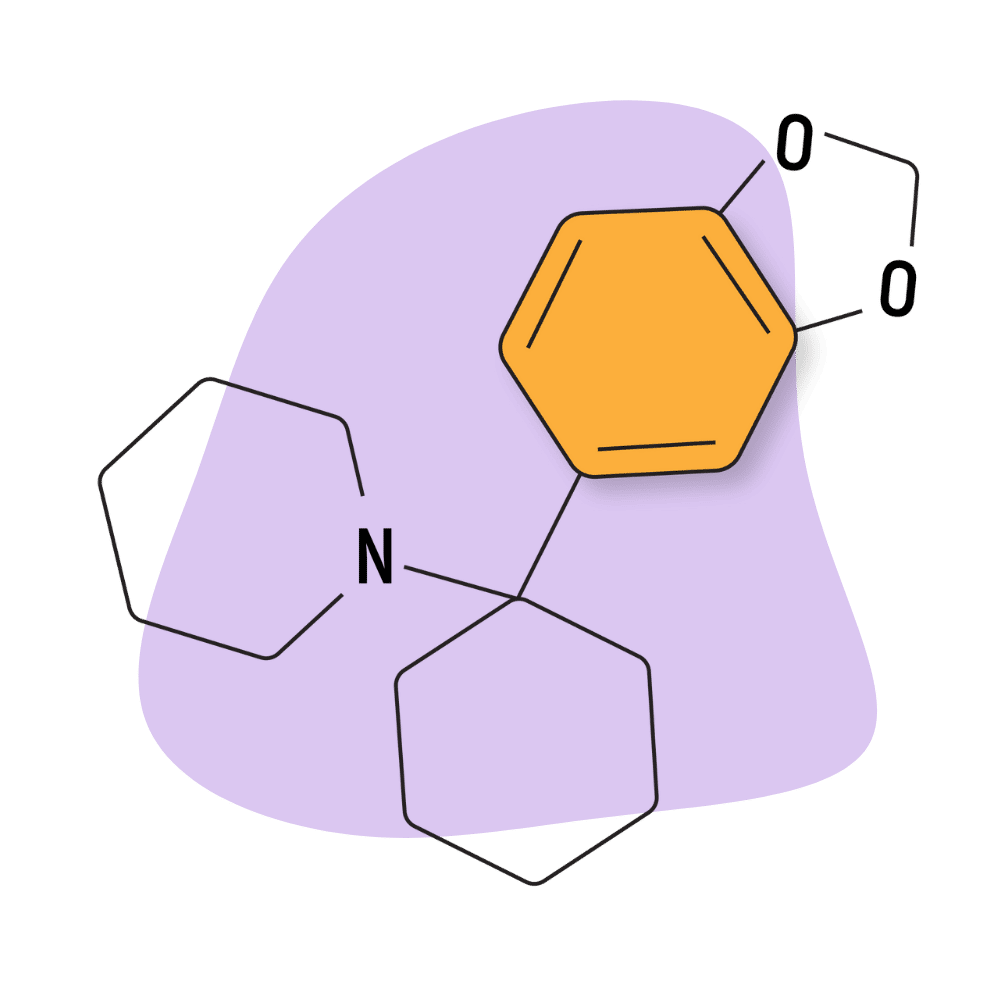 | 3′,4′-MD-PCP | Research Chemical 🧪 | 3 mg | 3–5 hours |
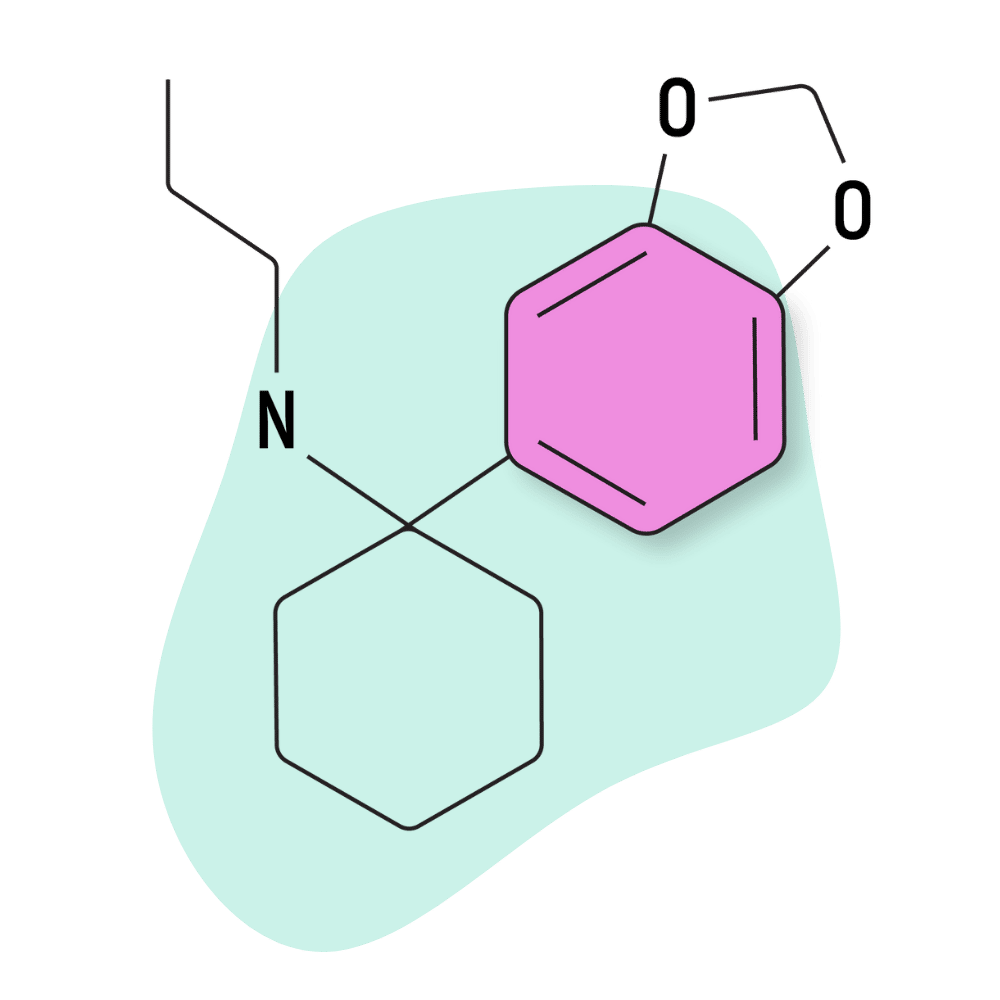 | 3′,4′-MD-PCPr | Research Chemical 🧪 | 3 mg | 3–5 hours |
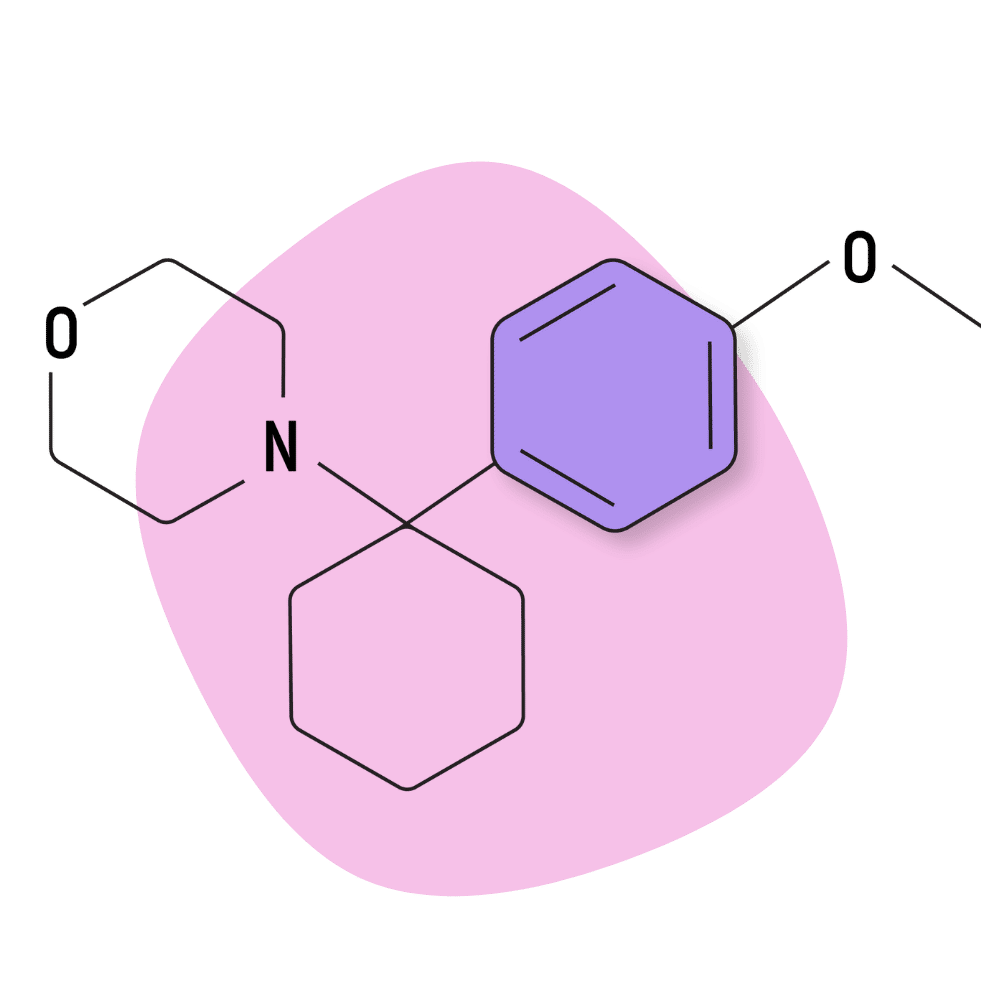 | 4′-MeO-PCMo | Research Chemical 🧪 | Unknown | Unknown |
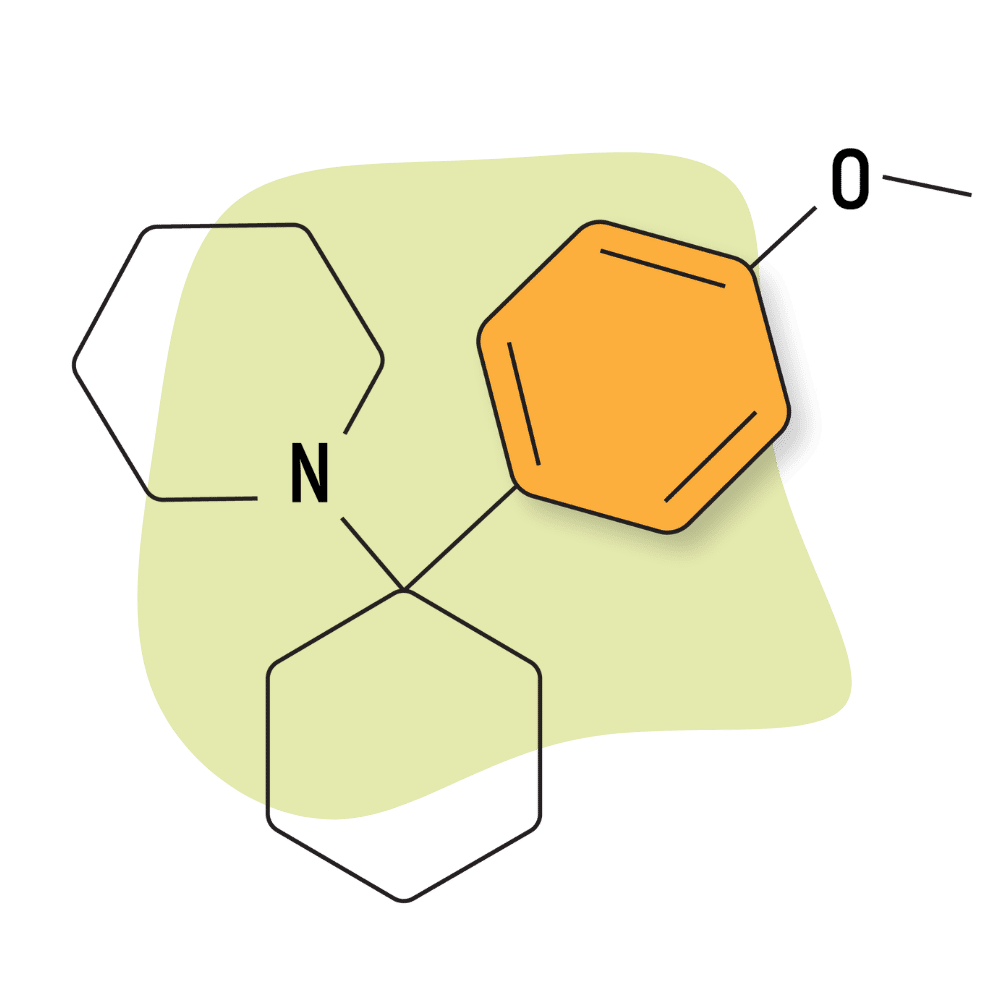 | 4′-MeO-PCP (Methoxydine) | Research Chemical 🧪 | 6 mg | Unknown |
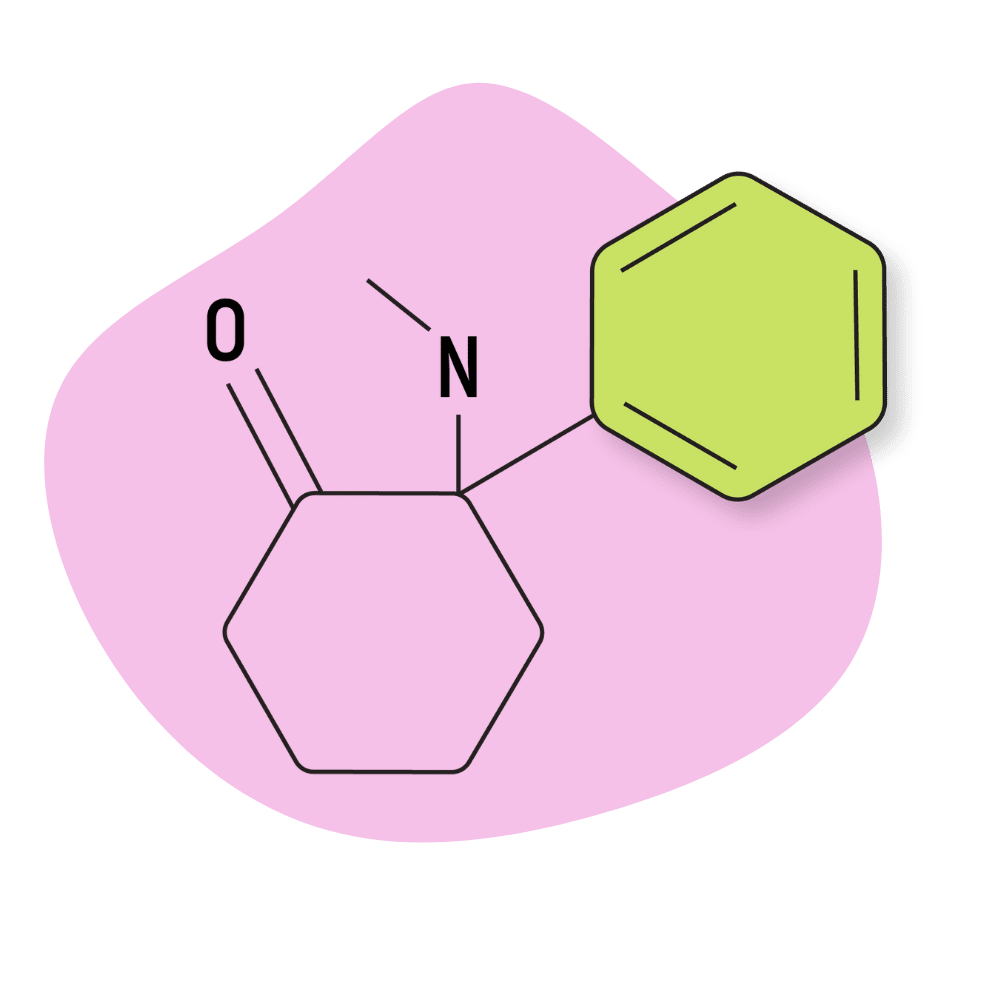 | DCK (Deschloroketamine, 2′-Oxo-PCM) | Research Chemical 🧪 | 5 mg | 4–6 hours |
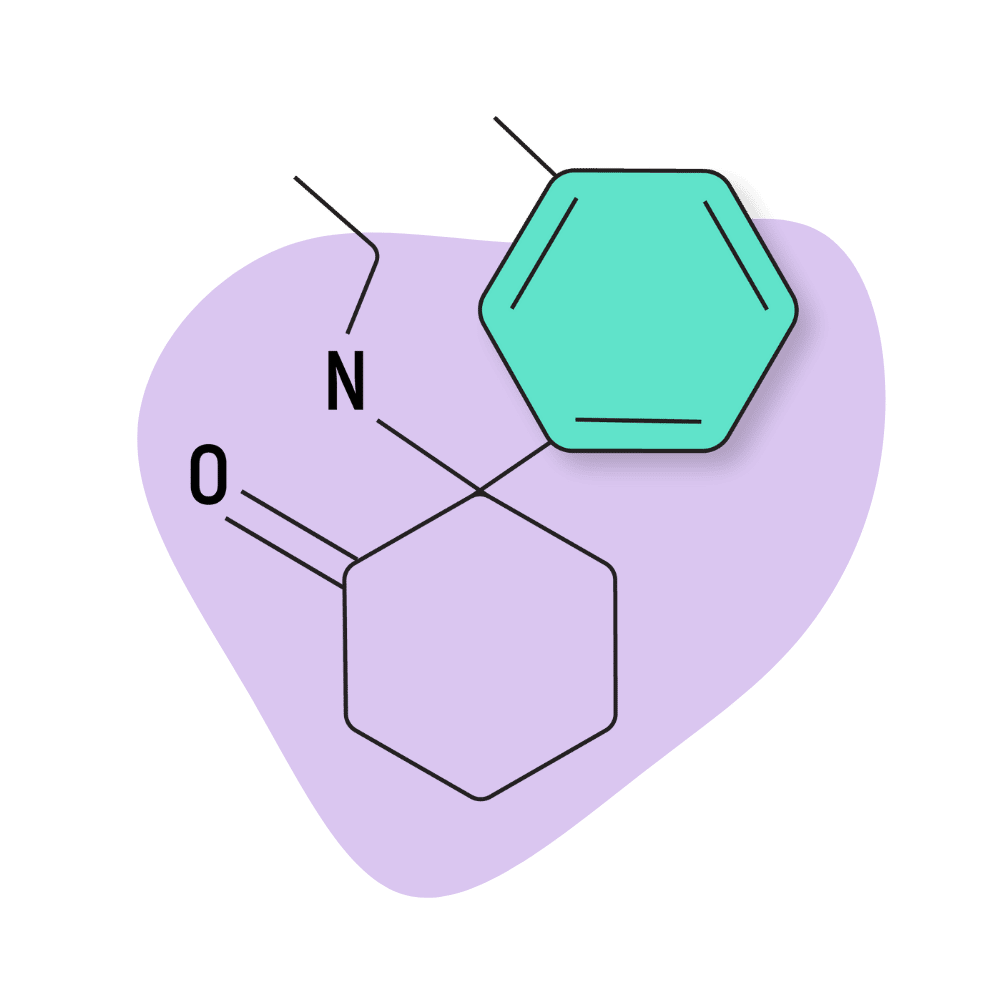 | DMXE (Deoxymethoxetamine, 3D-MXE) | Research Chemical 🧪 | 5 mg | 2–4 hours |
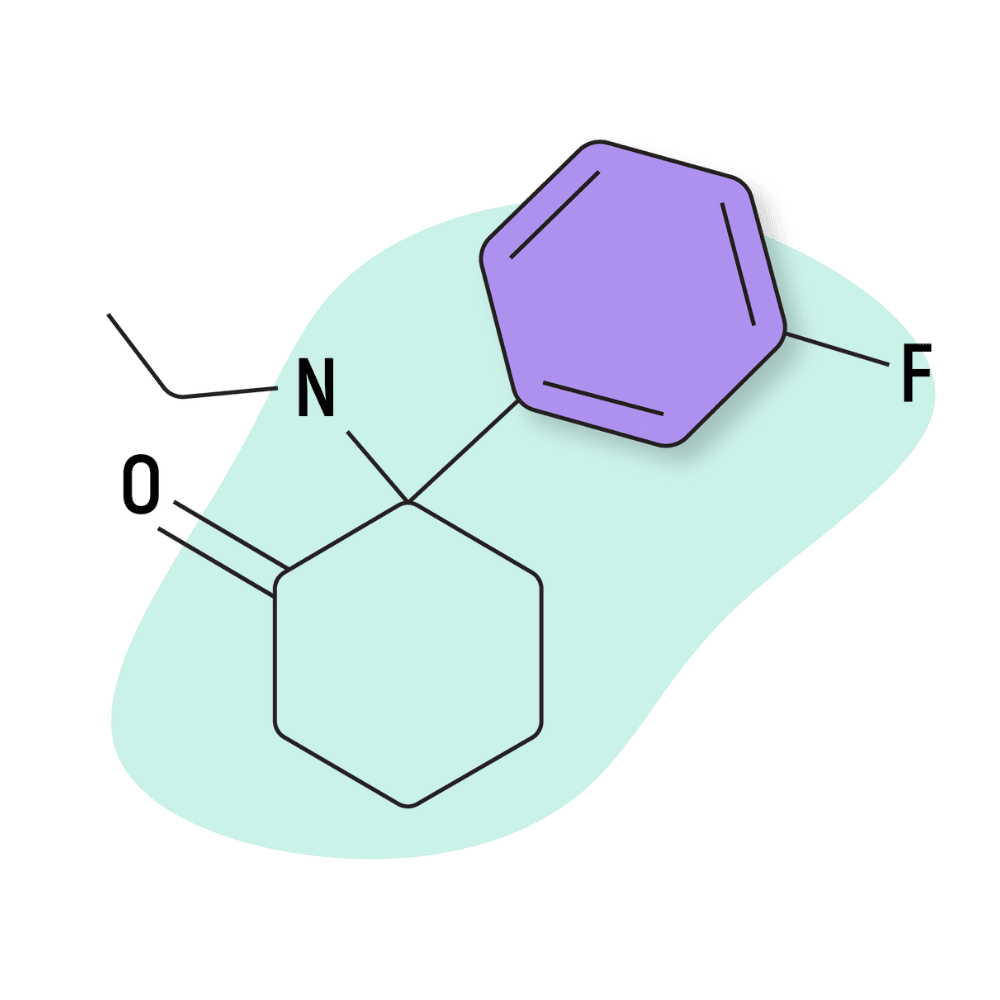 | FXE (Fluorexetamine) | Research Chemical 🧪 | 5 mg | 2–3 hours |
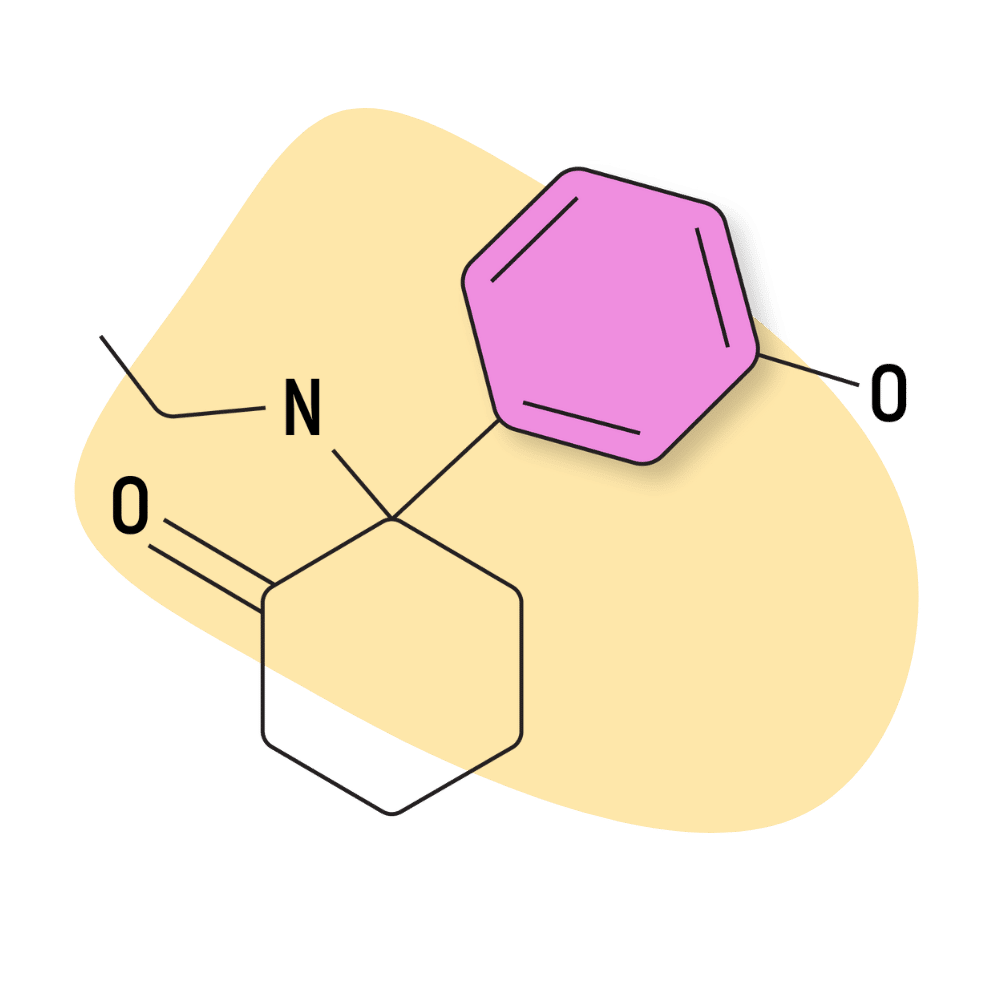 | HXE (Hydroxetamine) | Research Chemical 🧪 | 20 mg | 3–7 hours |
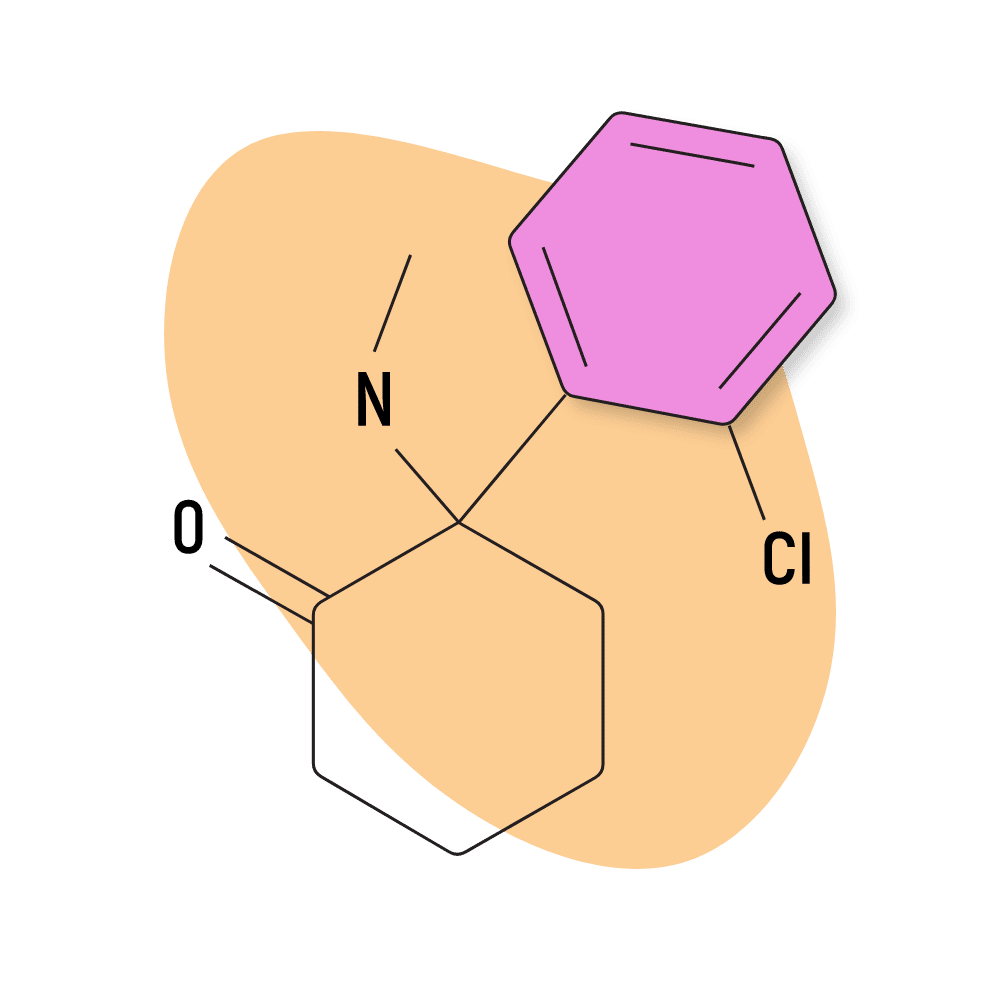 | Ketamine | Approved 💊 | 5 mg | 1–3 hours |
 | MXE (Methoxetamine) | Research Chemical 🧪 | 5 mg | 3–6 hours |
 | MXiPr (Methoxisopropamine) | Research Chemical 🧪 | 5 mg | 3–6 hours |
 | MXPr (Methoxpropamine) | Research Chemical 🧪 | 5 mg | 1–2 hours |
 | O-PCE (2-Oxo-PCE, Eticyclidone) | Research Chemical 🧪 | 1 mg | 3–6 hours |
 | PCE (Eticyclidine) | Research Chemical 🧪 | 1 mg | 4–7 hours |
 | PCP (Phencyclidine) | Discontinued | 1 mg | 4–8 hours |
 | TCP (Tenocyclidine) | Research Chemical 🧪 | 1 mg | Unknown |
 | Tiletamine | RC 🧪 | 3 mg | 3–6 hours |

2-BrDCK (Bromoketamine)
Bromoketamine (AKA 2-Br-2-Oxo-PCM, 2-Bromodeschloroketamine, or 2-BrDCK) is a structural analog of ketamine. The only difference is that the chlorine atom is replaced with a bromine atom.
This alteration produced very little if any, alteration to the subjective effects but significantly reduces the dissociative potency. Bromoketamine is considered one of the weakest members of the arylcyclohexylamines. Some users online report taking as much as 2000 mg (with tolerance) and only experiencing mild visual or auditory enhancement (doses this high are NOT recommended).
With that said, this compound is relatively new, so there aren’t many trip reports available to elucidate exactly how it compares to ketamine or other arylcyclohexylamine compounds.
Because of the reduced potency, this compound requires much higher doses than ketamine — leading to an increased risk of serious damage and pain to the nasal cavity (if snorted) and a higher risk of toxic metabolites damaging the kidneys and bladder.
As a result, most people who take this drug choose to inject it instead (IM), but even then, many users claim the effects aren’t nearly as enjoyable as other members of this class.
Most of the research involving this drug uses it as a control chemical for ketamine research [1,2]. There are no official studies examining the effects or safety of this drug specifically.
The receptor binding affinity for 2-BDCK has not yet been reported.
2-BrDCK Specs:
| Chemical Name | 2-Bromodeschloroketamine |
| Status | Research Chemical 🧪 |
| Duration of Effects | 1–3 hours |
| Estimated Threshold Dose | 150 mg |
| Common Dose | 200–500 mg |
| PubChem ID | 71588007 |
| CAS# | 120807-70-7 |

IUPAC Name: 2-(2-Bromophenyl)-2-(methylamino)cyclohexan-1-one
2-FDCK (2-Fluorodeschloroketamine)
2-FDCK is an analog of ketamine first described in a paper from 2014 by a group of researchers exploring the potential for new anesthetics based on various ketamine derivatives [3]. The drug started to appear on designer drug markets just 2 years later, in 2016.
Similar to ketamine, the effects of 2-FDCK are highly dose-dependent. Lower doses feel similar to intoxication from alcohol, but higher doses produce a profound hallucinogenic dissociative state (AKA a K-hole).
This compound features a fluorine molecule in the 2-position (rather than chlorine found in ketamine). This is believed to enhance the binding affinity to the NMDA receptors and therefore increase the potency of this drug. However, no official studies have been performed to confirm this is the case, and anecdotal reports are inconclusive.
2-FDCK Specs:
| Chemical Name | 2-Fluorodeschloroketamine |
| Status | Research Chemical 🧪 |
| Duration of Effects | 2–4 hours |
| Estimated Threshold Dose | 10 mg |
| Common Dose | 10–140 mg |
| PubChem ID | 13771618 |
| CAS# | 111982-50-4 |
IUPAC Name: 2-(2-Fluorophenyl)-2-methylamino-cyclohexanone
3,4-MD-PCE
3,4-MD-PCE shares similarities in structure with MDMA (3,4-methylenedioxy-methamphetamine) — which is essentially the 3,4-MD version of amphetamine. Despite the similarities in chemical structure, these molecules have very little in common. The base structure of MDMA is an amphetamine (stimulant), while the base structure of 3,4-MD-PCE is an arylcyclohexylamine (dissociative).
3,4-MD-PCE has shown a high binding affinity for the NMDA receptors (35 nM Ki) and low activity at the dopamine and norepinephrine transporters (>10,000 nM IC50)— suggesting a strong dissociative action above anything else. One study found this compound had moderate activity at the serotonin transporter (637 nM Ki), but this has not been confirmed.
3,4-MD-PCE Specs:
| Chemical Name | 3,4-methylenedioxy-eticyclidine |
| Status | Research Chemical 🧪 |
| Duration of Effects | 3–5 hours |
| Estimated Threshold Dose | 2 mg |
| Common Dose | 5–20 mg |
| PubChem ID | Not Available |
| CAS# | Not Available |
IUPAC Name: 1-(2H-1,3-Benzodioxol-5-yl)-N-ethylcyclohexan-1-amine
3,4-MD-PCMo
Like other members of the 3,4-MD- group, this compound has shown a much higher affinity for the NMDA receptors than other receptors, such as the dopamine, serotonin, and norepinephrine transporters. While the impacts of this remain inconclusive, it’s believed this effect gives this group of drugs a particularly strong dissociative quality and less euphoric, stimulant, and crossover effects than other arylcyclohexylamines.
With that said, the 3,4-MD-PCMo has the lowest activity at the NMDA receptors of the 3,4-MD- group at 425 nM (Ki). Comparatively, the binding affinity for related compounds is 62.8 nM (3,4-MD-PCP), 24.8 nM (3,4-MD-PCPr), and 35.5 nM (3,4-MD-PCE). The lower the number, the stronger a compound’s affinity for specific receptors.
3,4-MD-PCMo Specs:
| Chemical Name | 3,4-methylenedioxy-cyclohexylmorpholine |
| Status | Research Chemical 🧪 |
| Duration of Effects | Unknown |
| Estimated Threshold Dose | 4 mg |
| Common Dose | 5–40 mg |
| PubChem ID | Not Available |
| CAS# | Not Available |
IUPAC Name: 4-[1-(2H-1,3-Benzodioxol-5-yl)cyclohexyl]morpholine
3,4-MD-PCP
The 3,4-MD group consists of a methylenedioxy group attached to the 3 and 4 positions of a phenyl ring of the arylcyclohexylamine base structure. This is the same substitution used on the methamphetamine molecule to create MDMA, as well as other powerful amphetamine psychedelics like MDPV and methylone.
3,4-MD PCP analogs don’t possess any of the empathogenic qualities of MDMA.
All members of the 3,4-MD group are brand new to the market and have very little in the way of trip reports or formal research. It’s unclear exactly how this group compares to drugs like PCP, PCE, or ketamine — however, early reports suggest them to be very potent and dose-dependent. Lower doses are euphoric and mellow, while higher doses quickly become manic and overwhelming.
Reddit user Nervwing offers what is likely the best trip report currently available on this drug. This report used a dose of 40 mg 3,4-MD-PCP taken intranasally. In the report, Nervewing suggests the drug is a “unique dissociative” that delivers different experiences with every trip. The underlying theme of these trips was summarized as having “a sense of being transferred to an enhanced other self.”
The receptor binding affinity (Ki) for 3,4-MD-PCP at the NMDA receptors was reported at 62.8 nM (high affinity). This places it in third place for potency behind 3,4-MD-PCPr (25 nM) and 3,4-MD-PCE (3.5.nM). There have not been any studies comparing the binding affinity for dopamine, serotonin, or norepinephrine transporters to date.
3,4-MD-PCP Specs:
| Chemical Name | 3,4-methylenedioxy-phencyclidine |
| Status | Research Chemical 🧪 |
| Duration of Effects | 3–5 hours |
| Estimated Threshold Dose | 3 mg |
| Common Dose | 5–40 mg |
| PubChem ID | Not Available |
| CAS# | Not Available |
IUPAC Name: 1-[1-(2H-1,3-Benzodioxol-5-yl)cyclohexyl]piperidine
3,4-MD-PCPr
This obscure arylcyclohexylamine is rarely mentioned in the online literature. However, it was claimed by Dr. Jason Wallach (one of the leading researchers on the arylcyclohexylamine family) to have the highest selectivity for the NMDA receptor of any compound currently known [8].
In theory, this selectivity for NMDA receptors could result in a “pure” dissociative effect profile, free of the empathogenic, stimulant, and opiate-like qualities brought about by the activation of other neurotransmitter systems.
Despite these unique qualities, not much is known about the subjective effects of 3,4-MD-PCPr. It isn’t readily available, and few people appear to have tried this compound. There are a few trip reports available for a closely-related compound, 3,4-MD-PCP, many of which suggest it to be euphoric and “smooth” in lower doses, but the effects quickly turn manic and chaotic in higher doses.
The receptor affinity testing for 3,4-MD-PCPr reports Ki values of 24.9 nM at the NMDA receptors and virtually no activity at the serotonin or norepinephrine transporters. There’s conflicting evidence for the activity at the dopamine transporters, but leading arylcyclohexylamine researcher Dr. Wallach suggests mild activity in this transporter system.
3,4-MD-PCPr Specs:
| Chemical Nam | 3,4-methylenedioxy-phencyclamine |
| Status | Research Chemical 🧪 |
| Duration of Effects | 3–5 hours |
| Estimated Threshold Dose | 3 mg |
| Common Dose | 5–40 mg |
| PubChem ID | Not Available |
| CAS# | Not Available |
IUPAC Name: 1-(2H-1,3-Benzodioxol-5-yl)-N-propylcyclohexan-1-amine
3-Cl-PCP
3-Cl-PCP (AKA 3-Chloro-PCP or 3-Chlorophencyclidine) is an obscure dissociative psychedelic closely related to PCP. The difference between 3-Cl-PCP and PCP is the addition of a chlorine group to the phenyl ring structure. This alteration gives 3-Cl-PCP a distinct effect profile from PCP.
3-Cl-PCP is strongly stimulating and has a high likelihood of producing uncomfortable physical side effects — including muscle pain, runny nose, fatigue, increased heart rate, and stomach pain.
This compound has a poor safety profile compared to other members of its group, especially when used in higher doses. The effects are also much less predictable and dose-dependent — some users report feelings of dissociation and euphoria, while others report overstimulation and delirium.
It’s also recommended to avoid snorting this compound, as the crystals will cause extensive damage to the nasal cavity.
Because of how new this substance is, there’ve been a lot of reports of contaminated or adulterated batches. Most contain at least some PCP, but many contain unknown ingredients as well. This can make accurate dosing even more difficult and dangerous.
The receptor binding activity of 3-Cl-PCP has not yet been reported.
3-Cl-PCP Specs:
| Chemical Name | 3-Chlorophencyclidine |
| Status | Research Chemical 🧪 |
| Duration of Effects | 3–6 hours |
| Estimated Threshold Dose | 15 mg |
| Common Dose | 20–100 mg |
| PubChem ID | 125502037 |
| CAS# | 2201-32-3 |
IUPAC Name: 1-[1-(3-chlorophenyl)cyclohexyl]piperidine
3-F-PCP (3-Fluoro-PCP)
3-Fluoro-PCP is one of the lesser-known PCP analogs. It’s identical to 3-Cl-PCP, only instead of a chlorine group, it features a fluoride. In many cases, the addition of fluorine to psychoactive molecules makes them pass across the blood-brain barrier more easily — leading to significantly higher potency. This is the case with 3-F-PCP compared to 3-Cl-PCP, but it still appears to be less potent than PCP or 3-MeO-PCP.
Unfortunately, the addition of fluorine also makes this drug a bit more toxic — free fluorine molecules released as the drug breaks down can irritate the tissues and cause damage to the neurons.
The duration of action for this drug is suggested to be around 4 hours long, which is around the same duration as 3-MeO-PCP (its closest counterpart in terms of effects).
There are only a few good trip reports for this drug so far, and most of them suggest it’s somewhat milder than PCP and ketamine but also has a clearer, less “wonky” feeling to it.
3-Fluoro-PCP Specs:
| Chemical Name | 3-Fluoro-Phencyclidine |
| Status | Research Chemical 🧪 |
| Duration of Effects | 2–4 hours |
| Estimated Threshold Dose | 5 mg |
| Common Dose | 10–40 mg |
| PubChem ID | 398433 |
| CAS# | 89156-99-0 |
IUPAC Name: 1-[1-(3-fluorophenyl)cyclohexyl]piperidine
3-HO-PCE
This drug was first reported in 2012. It was developed by online enthusiasts exploring the production of new PCP analogs.
The drug company Eli Lilly briefly explored this compound as a potential medicine but quickly abandoned the project and redacted all published research the company submitted regarding the chemical.
The receptor Ki values for 3-HO-PCE are reported at 23 nM for the NMDAR, which would make it one of the strongest PCP analogs available today. However, the original source for this data could not be confirmed. Binding affinity for the NET, DAT, and SERT have not been tested.
3-HO-PCE Specs:
| Chemical Name | 3-Hydroxyeticyclidine |
| Status | Research Chemical 🧪 |
| Duration of Effects | 3–6 hours |
| Estimated Threshold Dose | 5 mg |
| Common Dose | 5–25 mg |
| PubChem ID | 137332171 |
| CAS# | 2243815-20-3 |
IUPAC Name: 3-[1-(Ethylamino)cyclohexyl]phenol
3-HO-PCP
3-HO-PCP was first reported on designer drug markets in 2009 but never achieved widespread popularity. This is likely because this drug carries a high risk of side effects like muscle pain, tension, and uncomfortable mental effects like confusion, irritability, and dizziness.
In terms of potency, 3-HO-PCP is stronger than PCP and about equal potency to 3-MeO-PCE. The effects are described as a combination of the dissociative qualities of PCP mixed with a notable opioid-like effect.
The NMDA receptor affinity of 3-HO-PCP is very high at around 30 nM. Some evidence 3-HO-PCP is active at the mu-opioid receptors as well [16].
Affinities for other receptors, including the serotonin transporter and norepinephrine transporter, appear negligible, and studies suggest a 3-fold reduction in dopamine reuptake capacity compared to PCP [17].
3-HO-PCP Specs:
| Chemical Name | 3-hydroxyphencyclidine |
| Status | Research Chemical 🧪 |
| Duration of Effects | 3–6 hours |
| Estimated Threshold Dose | 1 mg |
| Common Dose | 1–10 mg |
| PubChem ID | 133277 |
| CAS# | 79787-43-2 |
IUPAC Name: 3-[1-(Piperidin-1-yl)cyclohexyl]phenol
3-Me-PCE
The Me (methy[phenyl]) group of arylcyclohexylamines are relatively new additions to the market, with little to no information regarding subjective effects. There is, however, some early research exploring the binding affinities of these compounds that can help elucidate what their potency and effects indexes may present.
Based on preliminary testing, 3-Me-PCE appears to be the strongest of the PCP analogs with a methylphenyl aromatic ring substitution.
With that said, there are no established dosages or subjective effects comparisons for this particular compound. Some anecdotal reports suggest this compound has what was referred to as a “hard edge” — suggesting the effects of this drug are sharp and pronounced rather than smooth and flowy like some of the other PCP analogs.
Of the -Me- PCP analogs tested, most show a high affinity for the NMDA receptors (comparable to drugs like MXE and ketamine) and moderate activity at the serotonin receptors. 3-Me-PCE had the highest activity at the serotonin transporters at a Ki of 117 nM. The NMDA receptor affinity was reported at 43.1 nM.
3-Me-PCE Specs:
| Chemical Name | 3-methyl-phencyclidine |
| Status | Research Chemical 🧪 |
| Duration of Effects | Unknown |
| Estimated Threshold Dose | 2 mg |
| Common Dose | Unknown |
| PubChem ID | 154133693 |
| CAS# | Not Available |
IUPAC Name: N-Ethyl-1-(3-methylphenyl)cyclohexan-1-amine
3-Me-PCMo
Not much is known about this drug or how it compares to other PCP analogs. However, based on some early testing and correlation with other PCMo-based compounds, it appears this drug is one of the weakest members of the -Me- group.
The binding affinity for 3-Me-PCMo was reported at 201.8 nM for the NMDA receptors — which would make it roughly 25% as potent as 3-Me-PCE. This data is based purely on binding affinity, which doesn’t always correlate with how the drug interacts inside a living human.
All information about this drug at the moment is purely speculative, and it doesn’t appear this compound is widely available on the designer drug market.
3-Me-PCMo Specs:
| Chemical Name | 3-methyl-phenylcyclohexyl-morpholine |
| Status | Research Chemical 🧪 |
| Duration of Effects | Unknown |
| Estimated Threshold Dose | Unknown |
| Common Dose | Unknown |
| PubChem ID | 137332197 |
| CAS# | Not Available |
IUPAC Name: 4-[1-(3-Methylphenyl)cyclohexyl]morpholine
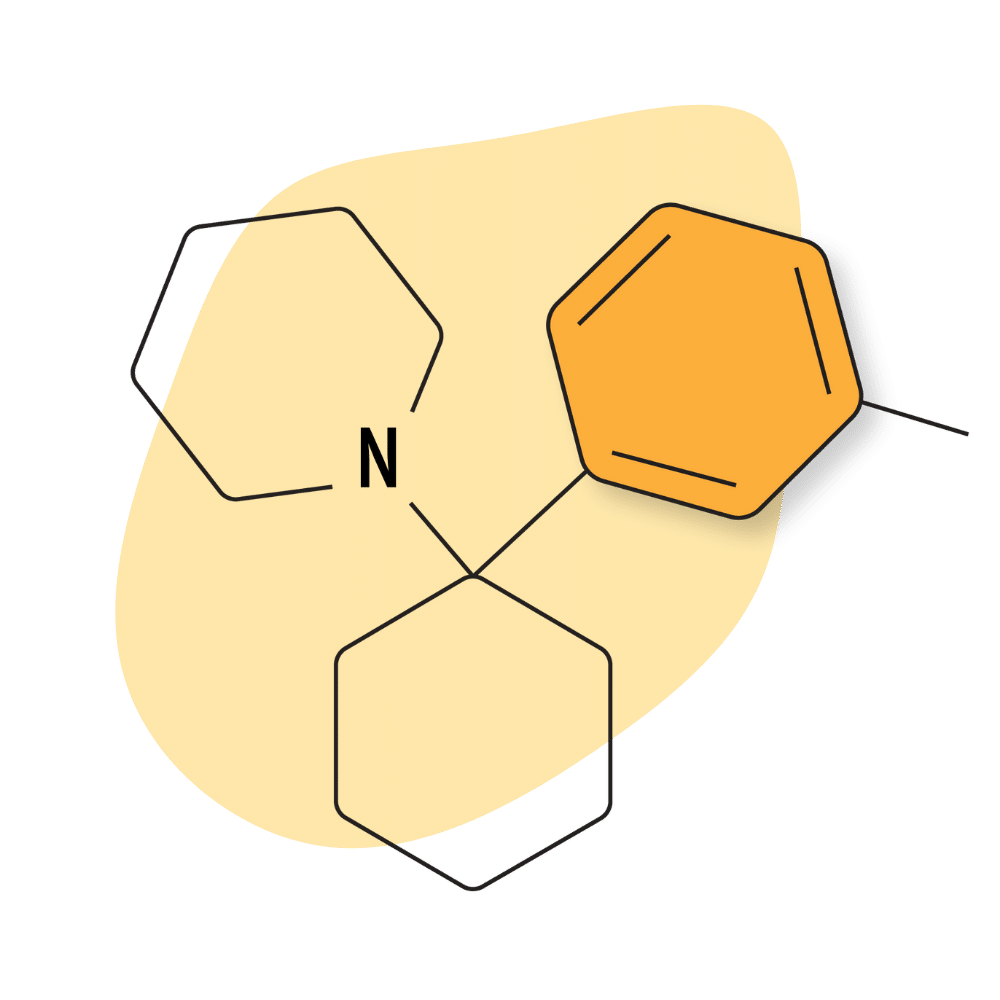
3-Me-PCP
3-Me-PCP was first synthesized in the 1960s [14] but didn’t become popular until around 2020, when it first started appearing on online designer drug marketplaces.
The effects of 3-Me-PCP are very lucid — users feel as though they’re in some sort of middle-ground between dreaming and reality. This comes with a layer of confusion and haziness some people find uncomfortable.
Despite the lucidity, 3-Me-PCP is quite stimulating — it causes the mind to feel like it’s racing which makes it harder to react to events occurring in the outside world. It’s common for people who take this drug to feel like they’re operating on a different timeline than everybody else (time is either going too fast or too slow). All of this makes it very difficult to interact with others or get anything done while using this substance, even with relatively low dosing. As a result, most consider this drug one of the least “functional” of the arylcyclohexylamines.
Another common theme in the trip reports for this substance is the development of rapidly increasing thought loops that tend to repeat themselves over and over again. This can give the drug a manic or chaotic nature.
Lower doses of this drug (2 or 3 mg) are markedly euphoric and stimulating with lots of interesting visuals. True hallucinations and dissociation usually don’t begin until the 5 or 6 mg mark for those without tolerance to PCP analogs.
The receptor Ki values for 3-Me-PCP have not yet been reported, but it’s suspected to have a high binding affinity for both the NMDA receptors and the norepinephrine transporter.
3-Me-PCP Specs:
| Chemical Name | 3-methyl-phencyclidine |
| Status | Research Chemical 🧪 |
| Duration of Effects | Unknown |
| Estimated Threshold Dose | 2 mg |
| Common Dose | 3–15 mg |
| PubChem ID | 611899 |
| CAS# | 2201-30-1 |
IUPAC Name: 1-[1-(3-methylphenyl)cyclohexyl]piperidine
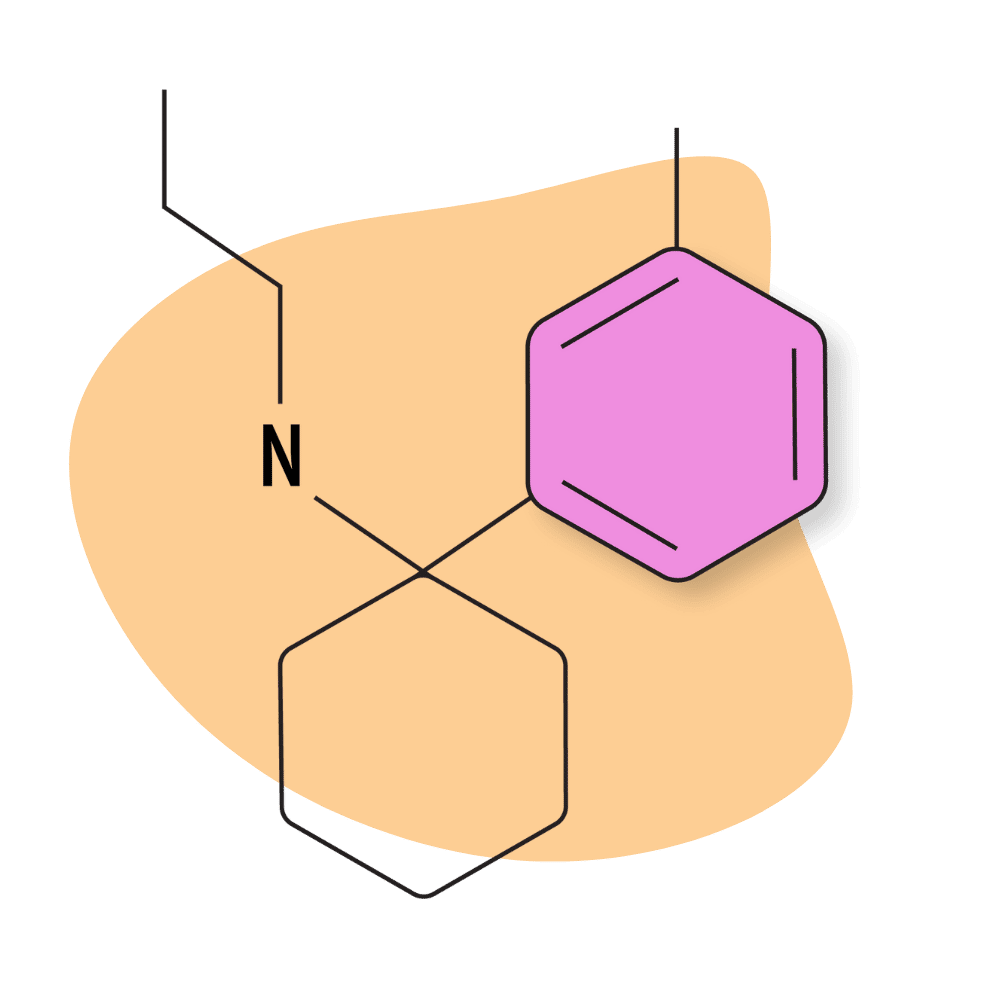
3-Me-PCPr
There’s virtually no mention of this drug on designer drug forums, and testing on this compound is scant.
However, there are a few insights we can derive from some of the early testing on this compound. For example, the PCPr group is known for being some of the most potent PCP analogs available. Receptor affinity testing places the Ki values for 3-Me-PCPr at around 53.5 nM, which could mean this compound is comparable in strength (if not slightly stronger) than PCP.
Related members of the PCPr group have even higher potency, with Ki values of around 18 nM and 25 nM at the NMDAR for 3,4-MD-PCPr and 3-MeO-PCPr, respectively.
This compound appears to be less specific for the NMDA receptors than other PCP analogs — suggesting unpredictable effects on mood, memory, and other areas of cognition. Preliminary receptor testing reports 113 nM for the DAT (moderate activity), 853 nM for the SERT (moderate activity), and 264 nM for the NET (moderate activity).
3-Me-PCPr Specs:
| Chemical Name | 3-methyl-phencyclamine |
| Status | Research Chemical 🧪 |
| Duration of Effects | Unknown |
| Estimated Threshold Dose | 1 mg |
| Common Dose | Unknown |
| PubChem ID | Not Available |
| CAS# | Not Available |
IUPAC Name: 1-(3-Methylphenyl)-N-propylcyclohexan-1-amine
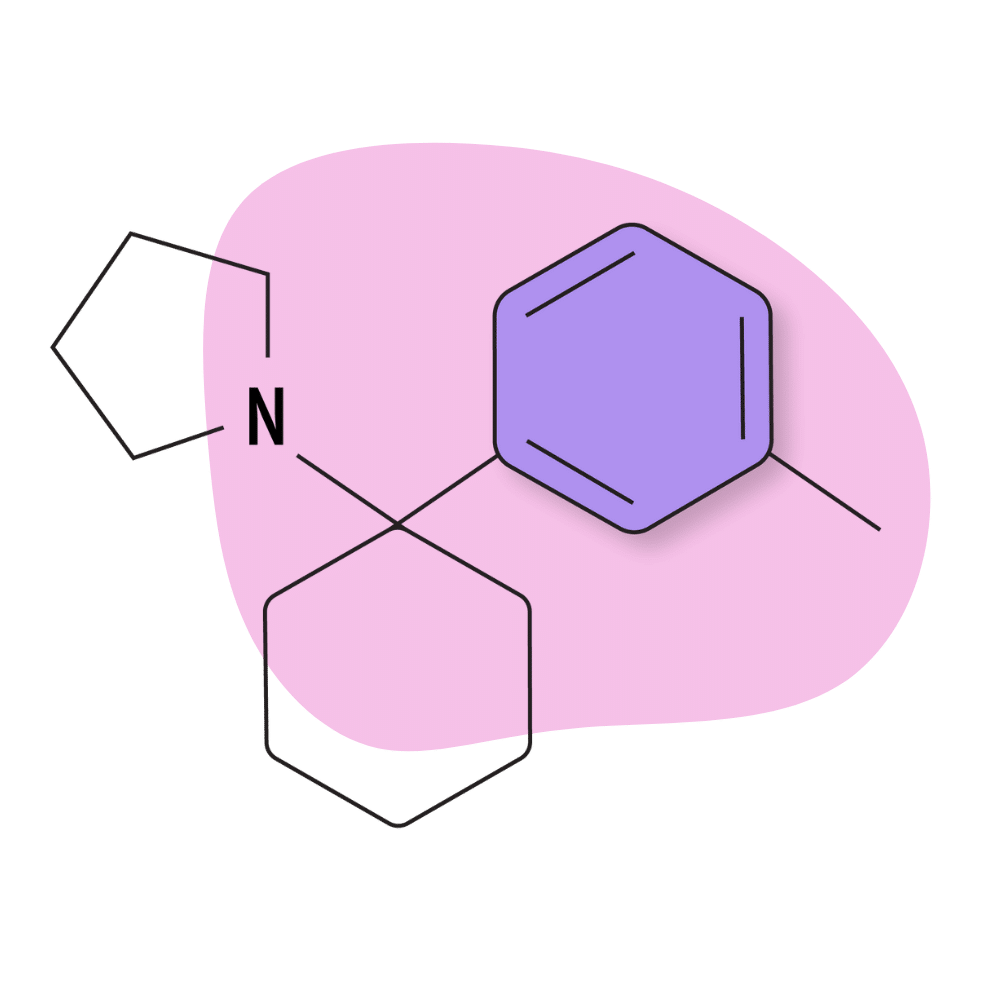
3-Me-PCPy
3-Me-PCPy is a lesser-known member of the arylcyclohexylamine family of drugs featuring pyrrolidine as the amide group (abbreviated as Py).
This compound acts as an NMDA receptor antagonist and triple-reuptake inhibitor — blocking the reuptake of dopamine, serotonin, and norepinephrine [18].
There aren’t many trip reports available for this drug, but those available suggest it’s a milder version of 3-MeO-PCP. It’s euphoric and stimulating — producing enhanced feelings of confidence and perceived mental clarity.
Like many other PCP analogs, this sense of clarity is synthetic — users often feel as though they can see things for what they really are, but their observations and “discoveries” are in fact delusional interpretations of reality. These depictions often wax and wane during the trip — users feel moments of clarity followed by moments of confusion and mania. This effect can sometimes persist for several hours or days after the effects of the drug wear off.
3-Me-PCPy Specs:
| Chemical Name | 3-methyl-rolicyclidine |
| Status | Research Chemical 🧪 |
| Duration of Effects | 3–6 hours |
| Estimated Threshold Dose | 3 mg |
| Common Dose | 3–15 mg |
| PubChem ID | 137332188 |
| CAS# | 1622348-63-3 |
IUPAC Name: 1-[1-(3-Methoxyphenyl)cyclohexyl]-pyrrolidine
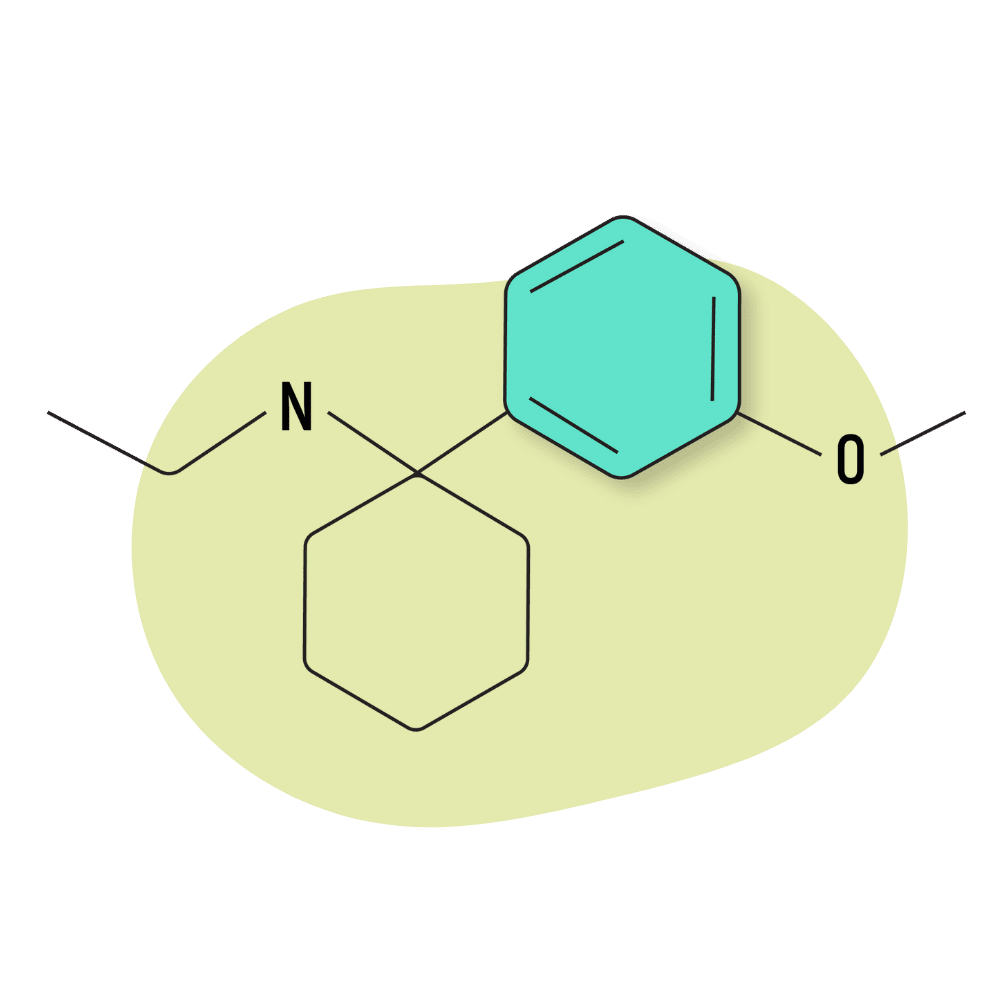
3-MeO-PCE
3-MeO-PCE is an analog of MXE (methoxetamine) — one of the more popular dissociative drugs in the arylcyclohexylamine family. Its effects are stimulating in lower doses and dissociative and hallucinogenic in higher doses.
However, tread cautiously with this one — the effects of 3-MeO-PCE are often reported as being more damaging to both the mind and body than other arylcyclohexylamines.
This compound is stronger than PCP and 3-MeO-PCP but tends to kick in more slowly (1–2 hours onset with oral use and up to 30 minutes insufflated).
Many users advise against using this drug because of its higher affinity for inducing mania than other PCP analogs. The mental stimulation of this drug can be intense — causing users to get stuck in aggressive delusional thought loops, sometimes lasting several hours at a time.
For this reason, many users report mixing it with a more sedative dissociative like DCK or ketamine or alongside a mild benzodiazepine to help offset this effect.
Even after the effects of this drug wear off, it’s common for users to experience difficulty falling asleep. Hangovers are also common with this compound — symptoms include apathy, difficulty organizing thoughts, and mild inebriated feeling.
Reports of this substance date back to at least 1979 [15], but it didn’t become popular as a research chemical until around 2010, as reported by the EMCDDA.
The receptor testing (Ki values) for 3-MeO-PCE are reported at 30.4 nM for the NMDAR, 1,528 nM for the DAT, 1,528 nM for the NET, and 136 nM for the SERT. This drug has a stronger serotonergic action than most other compounds in this class.
3-MeO-PCE Specs:
| Chemical Name | 3-methoxy-eticyclidine |
| Status | Research Chemical 🧪 |
| Duration of Effects | 3–5 hours |
| Estimated Threshold Dose | 3 mg |
| Common Dose: | 5–20 mg |
| PubChem ID | 57461569 |
| CAS# | 1364933-80-1 |
IUPAC Name: N-Ethyl-1-(3-methoxyphenyl)cyclohexan-1-amine
3-MeO-PCMe
This compound is one of several arylcyclohexylamines featuring a methylamino group. Related compounds include 2-BrDCK, 2-FDCK, DCK, and ketamine.
The subjective effects of 3-MeO-PCMe are not well defined, but based on potency testing; it’s likely this compound exerts a similar dissociative effect to ketamine but with a unique nature of effects due to differences in activity on dopamine, serotonin, and norepinephrine transporters.
The receptor Ki values for 3-MeO-PCMe are reported at 145.1 nM for the NMDAR (high), 3,238 nM for the DAT (low), 3,238 nM for NET (low), and 1,368 nM for the SERT (low).
3-MeO-PCMe Specs:
| Chemical Name | 3-methoxy-phenylcyclohexylamine |
| Status | Research Chemical 🧪 |
| Duration of Effects | Unknown |
| Estimated Threshold Dose | 8 mg |
| Common Dose | 10–20 mg |
| PubChem ID | Not Available |
| CAS# | Not Available |
IUPAC Name: 1-(3-Methoxyphenyl)-N-methylcyclohexan-1-amine
3-MeO-PCMo
3-MeO-PCMo was first reported by the EMCDDA in 2015, but its precursor, PCMo, was described in a patent dating back to 1954. This could make PCMo older than PCP.
This compound was made in the UK in an attempt to circumvent legislation banning other members of the arylcyclohexylamine family.
3-MeO-PCMo is the least potent member of the PCP-analogs — with common dosages ranging from 100–500 mg (compared to 1–10 mg for PCP). It was found to have a comparable binding affinity for the NMDA receptors as ketamine and about 12-fold lower activity than PCP [6].
3-MeO-PCMo features a morpholine group as the amide (Mo). There are several different analogs of PCMo available today, including 2-MeO-PCMo, 4-MeO-PCMo, 3-Me-PCMo, and 3,4-methelendioxy-PCMo.
The receptor Ki values for 3-MeO-PCMo are reported at 252.9 nM for the NMDA receptors. Both 2-MeO-PCMo and 3-MeO-PCMo had a higher affinity for the serotonin transporters over dopamine transporters, no activity at any of the opioid receptors, and little to no activity at the sigma 1 and sigma 2 receptors.
3-MeO-PCMo Specs:
| Chemical Name | 3-methoxy-phenylcyclohexyl-morpholine |
| Status | Research Chemical 🧪 |
| Duration of Effects | Unknown |
| Estimated Threshold Dose | 50 mg |
| Common Dose | 100–500 mg |
| PubChem ID | 132605908 |
| CAS# | 138873-80-0 |
IUPAC Name: 4-[1-(3-methoxyphenyl)cyclohexyl]morpholine
3-MeO-PCP
3-MeO-PCP (AKA 3-Methoxyphencyclidine) was first synthesized in 1979 as part of some early research comparing the effects of PCP and its analogs. It didn’t enter the market as a research chemical until many years later, in 2011.
This compound is very potent, even stronger than PCP, 2-MeO-PCP, and 4-MeO-PCP.
Unfortunately, it also has a high likelihood of inducing hypomanic states. Some people like this effect, but it can be scary for others. People looking for a “chill” arylcyclohexylamine should avoid this one. Even low doses appear to carry a strong sense of delusional thought patterns and stimulation.
Some early reports suggested this drug also had opioid-like activity — users would feel physically numb and relaxed, with an underpinning sense of euphoria. However, researchers have tested this theory and compared the affinity of this drug on the μ-, δ-, or κ- opioid receptors but found no activity at all [11]. It’s likely that the strong dissociative action is what is offering an almost opioid-like quality — users feel disconnected from the body, which inherently offers some painkilling effects.
3-MeO-PCP was found to have a high binding affinity (Ki) for the MK-801 binding site of the NMDA receptors (20–38.1 nM) and sigma 1 receptors (42 nM), moderate affinity for the serotonin transporter (216–1571 nM), low activity on the norepinephrine transporter (1,808 nM), and very low or no activity at the sigma 2 receptors and dopamine transporters [5,11].
3-MeO-PCP Specs:
| Chemical Name | 3-Methoxyphencyclidine |
| Status | Research Chemical 🧪 |
| Duration of Effects | 3–5 hours |
| Estimated Threshold Dose | 1 mg |
| Common Dose | Unknown |
| PubChem ID | 11778080 |
| CAS# | 72242-03-6 |
IUPAC Name: 1-[1-(3-methoxyphenyl)cyclohexyl]-piperidine
3-MeO-PCPy
This compound is a good example of a drug developed by clandestine drug manufacturers rather than scientific researchers or pharmaceutical companies. There are a lot of highly skilled and passionate chemists operating in the psychedelic community who have worked together on several occasions to bring new psychoactive substances to the table. 3-MeO-PCPy is just one example.
There aren’t many trip reports available for this compound, and official research has only skimmed the surface (in vitro metabolic and receptor binding studies) [26].
Most trip reports involving this substance suggest it’s more stimulating than most arylcyclohexylamines which is likely due to its potent triple-reuptake activity. 3-MeO-PCPy inhibits the reuptake of dopamine, serotonin, and norepinephrine to a significant degree.
More specifically, the receptor Ki values for 3-MeO-PCPy are reported at 22.3 nM for the NMDAR, 96 nM for the DAT, 96 nM for the NET, 11 nM for the SERT. All of these values indicate much higher potency than the average member of the arylcyclohexylamine family. This is a trait found in most PCPy analogs.
3-MeO-PCPy Specs:
| Chemical Name | 3-methoxy-rolicyclidine |
| Status | Research Chemical 🧪 |
| Duration of Effects | Unknown |
| Estimated Threshold Dose | 2 mg |
| Common Dose | 5–20 mg |
| PubChem ID | Not Available |
| CAS# | Not Available |
IUPAC Name: 1-[1-(3-Methoxyphenyl)cyclohexyl]pyrrolidine
4-MeO-PCMo
4-MeO-PCMo isn’t a popular research chemical, likely because of its relatively low potency compared to other members of the arylcyclohexylamine family.
The qualitative effects of this drug are not well defined, and there aren’t many good trip reports available for this drug online.
Preliminary testing suggests the Ki receptor binding affinity of this drug is around 2118 nM for the NMDA receptors (low activity) and negligible for the dopamine, serotonin, and norepinephrine transporters.
4-MeO-PCMo Specs:
| Chemical Name | 4-methoxy-phenylcyclohexyl-morpholine |
| Status | Research Chemical 🧪 |
| Duration of Effects | Unknown |
| Estimated Threshold Dose | Unknown |
| Common Dose | Unknown |
| PubChem ID | 137332210 |
| CAS# | 2201-35-6 |
IUPAC Name: 4-[1-(4-Methoxyphenyl)cyclohexyl]morpholine
4-MeO-PCP (Methoxydine)
4-MeO-PCP was one of the first compounds to enter the market during the new wave of online designer drug markets. The chemical structure was just different enough for vendors to sell the material without directly breaking any laws.
This compound was first sold online in 2008, and its success sparked an entire industry of designer PCP and ketamine analogs which continue to come out every year [4]. With that said, the first publication of 4-MeO-PCP synthesis dates back to the 1960s [14]. It remained on the shelf for decades until someone realized they could sell the compound pseudo-legally as a PCP alternative.
4-MeO-PCP is less potent than PCP and 3-MeO-PCP but still carries substantial dissociative and hallucinogenic properties in doses of around 40 mg.
The receptor Ki values for 4-MeO-PCP are reported at 620 nM for the NMDAR, 1,811 for the DAT, 1,811 for the NET, and 900 for the SERT.
4-MeO-PCP Specs:
| Chemical Name | 4-Methoxyphencyclidine |
| Status | Research Chemical 🧪 |
| Duration of Effects | Unknown |
| Estimated Threshold Dose | 6 mg |
| Common Dose | 20–40 mg |
| PubChem ID | 15100753 |
| CAS# | 2201-35-6 |
IUPAC Name: 1-[1-(4-Methylphenyl)cyclohexyl]piperidine
DCK (Deschloroketamine, 2′-Oxo-PCM, O-PCM, DXE)
DCK is closely related to ketamine — the only difference is that DCK lacks the chlorine functional group present in ketamine. This subtle difference makes a noticeable difference in terms of the effects. Both drugs share similar dissociative and sedative qualities but are undeniably different in how they feel.
DCK is stronger than ketamine and has a stronger visual component. It also lasts longer than ketamine (up to 6 hours compared to 2 or 3) and appears to have a lower chance of causing bladder damage. The reports on this drug are inconclusive; many people claim it to be only mildly active, even in high doses. Others suggest it’s much stronger than the equivalent dose of ketamine. There appears to be a genetic component to this drug that causes different people to react differently to the drug.
Despite the differences, the general consensus is that DCK is the closest alternative to ketamine — especially compared to the more stimulating arylcyclohexylamines like PCP or O-PCE.
This compound has a strongly lucid effect in lower doses. Users experience changes in visual perception, mild euphoria, dissociation, and dream-like states of consciousness. It can make users feel confused or disoriented, especially with regard to timeframes and complex decision-making. Higher doses can induce K-hole levels of dissociation.
DCK is stronger and longer-lasting than ketamine and appears to be less prone to causing bladder damage. This is due to the fact that users need a lower dose of DCK to produce similar effects — which means less toxic metabolites end up in the urine.
To be clear, DCK still produces the same bladder issues as ketamine (bladder pain, blood in the urine, and incontinence) — it just takes longer to appear in most people.
The receptor binding affinity for DCK on the NMDAR, DAT, SERT, and NET has not been reported despite the relative popularity of this compound.
DCK Specs:
| Chemical Name | Deschloroketamine |
| Status | Research Chemical 🧪 |
| Duration of Effects | 4–6 hours |
| Estimated Threshold Dose | 5 mg |
| Common Dose | 5–25 mg (Oral & Insufflated) |
| PubChem ID | 437168 |
| CAS# | 7063-30-1 |
IUPAC Name: 2-Phenyl-2-(methylamino)cyclohexanone
DMXE (Deoxymethoxetamine, 3D-MXE)
The effects of DMXE (deoxymethoxetamine) are most similar to MXE but a bit weaker and “smoother.” MXE has a higher tendency to produce scary or uncomfortable dissociated states — users feel confused, disorientated, and afraid. DMXE can produce the same but is more likely to induce all the positive qualities of these compounds — a “wonky” feeling of dissociation from the body, trippy hallucinations, and feelings of calmness and euphoria.
With that said, DMXE is not widely available, and the trip reports covering the subjective effects of this drug are varied. The general consensus is that it’s weaker and smoother than other MXE, PCP, and PCE analogs.
The receptor Ki values for DMXE have not been reported.
DMXE Specs:
| Chemical Name | Deoxymethoxetamine |
| Status | Research Chemical 🧪 |
| Duration of Effects | 2–4 hours |
| Estimated Threshold Dose | 5 mg |
| Common Dose | 20–60 mg |
| PubChem ID | 157010705 |
| CAS# | 2666932-45-0 |
IUPAC Name: 2-(ethylamino)-2-(3-methylphenyl)cyclohexan-1-one
FXE (Fluorexetamine)
The structure of FXE closely resembles ketamine — the difference is that a chlorine atom in ketamine is swapped for fluorine. The amine group also features one extra carbon in the chain (methylamino vs. ethylamino).
The effects of FXE are described as being virtually identical to ketamine but longer lasting and slightly more euphoric. It produces a strong body high, even in low doses, yet many consider it a great “functional” dissociative. Some even describe it as a “social dissociative,” similar to low-dose ketamine or some of the PCP analogs. The dose of FXE also tends to be more forgiving than other, more potent arylcyclohexylamines like 3-MeO-PCPy or 2-FDCK.
The line between a “social” dose and K-hole experience isn’t as subtle with FXE as it is with drugs like ketamine, MXE, PCE, or DCK — especially when using the drug via IM injections.
Some reports suggest FXE is less likely to lead to the bane of arylcyclohexylamine abuse and bladder damage, but this has never been confirmed. Its likely FXE is converted into the same toxic metabolites as ketamine but takes longer to develop because of how small the dose is.
The receptor Ki values for FXE have not been reported.
FXE Specs:
| Chemical Name | Fluorexetamine |
| Status | Research Chemical 🧪 |
| Duration of Effects | 2–3 hours |
| Estimated Threshold Dose | 5 mg |
| Common Dose | 10–60 mg |
| PubChem ID | 137332183 |
| CAS# | Not Available |
IUPAC Name: 2-(ethylamino)-2-(3-fluorophenyl)cyclohexan-1-one
HXE (Hydroxetamine)
HXE (AKA hydroxetamine, 3′-hydroxy-2-oxo-PCE, and O-desmethylmethoxetamine) is one of the active metabolites of another dissociative arylcyclohexylamine, methoxetamine (MXE) [20]. Structurally, the difference between HXE and MXE is a replacement of the hydroxy group (HXE) for the methoxy group (MXE).
Online vendors started selling HXE around 2018, shortly after it was discovered to have comparable dissociative and hallucinogenic properties to its parent compound.
The effects of HXE differ from MXE in that it’s viewed as “heavier” than MXE (referring to its sedative, cognitive suppressing activity). MXE is already considered more sedating than other arylcyclohexylamines and tends to cause users to feel confused or groggy.
Several prominent users on the /r/researchchemicals subreddit suggest this compound is “aquatic” in nature. The metaphor is that HXE takes users to an abysmal zone where it’s dark, vast, and pressurized. HXE makes it hard to move and think clearly but lacks the disturbing visuals that are common with MXE.
The receptor Ki values for HXE have not yet been reported.
HXE Specs:
| Chemical Name | Hydroxetamine |
| Status | Research Chemical 🧪 |
| Duration of Effects | 3–7 hours |
| Estimated Threshold Dose | 20 mg |
| Common Dose | 40–120 mg |
| PubChem ID | 472175405 |
| CAS# | 1620054-73-0 |
IUPAC Name: 2-(ethylamino)-2-(3-hydroxyphenyl)cyclohexan-1-one
Ketamine
Out of all the arylcyclohexylamines in existence, ketamine is, by far, the most well-used and well-researched. It’s also the only group member still used today in mainstream medicine as both an anesthetic and psychedelic-assisted psychotherapy. Ketamine is considered a breakthrough therapy for treating clinical depression. It’s also used to help restore damaged neurons following a stroke or head injury.
Ketamine was the third arylcyclohexylamine created by the pharmaceutical company Parke-Davis in the 1950s and 60s. PCP came first but was discontinued because of a problematic side effect called “emergence delirium.” PCE was developed next but never made it to market because the effects of the newest molecule, ketamine, were deemed superior.
The effects of ketamine change depending on the dose. In lower doses, ketamine is very similar to alcohol — producing a feeling of dissociation, as though the body is on autopilot.
Users also report enhanced sociability, mild euphoria, and a reduction of anxiety or worries.
In higher doses, ketamine becomes much more dissociative and hallucinogenic. At K-hole levels (~100 mg orally/insufflated), ketamine is a formidable psychedelic — comparable to such drugs as DMT or salvia, but with a distinctly dissociative nature.
Ketamine causes users to experience consciousness without a body. All sensory and bodily information is cut off, but users remain fully in control of the body (unlike benzodiazepines), and respiration isn’t suppressed (unlike opiates).
The receptor Ki values for ketamine are reported at 323.9 nM on the NMDA receptors, which places is about average in terms of potency. Ketamine also interacts with the dopamine, serotonin, and norepinephrine transporters, opioid receptors, and sigma 1 and sigma 2 receptors.
Ketamine Specs:
| Chemical Name | Ketamine |
| Status | Research Chemical 🧪 |
| Duration of Effects | 1–3 hours |
| Estimated Threshold Dose | 5 mg |
| Common Dose | 20–80 mg |
| PubChem ID | 3821 |
| CAS# | 6740-88-1 |
IUPAC Name: (RS)-2-(2-Chlorophenyl)-2-(methylamino)cyclohexanone
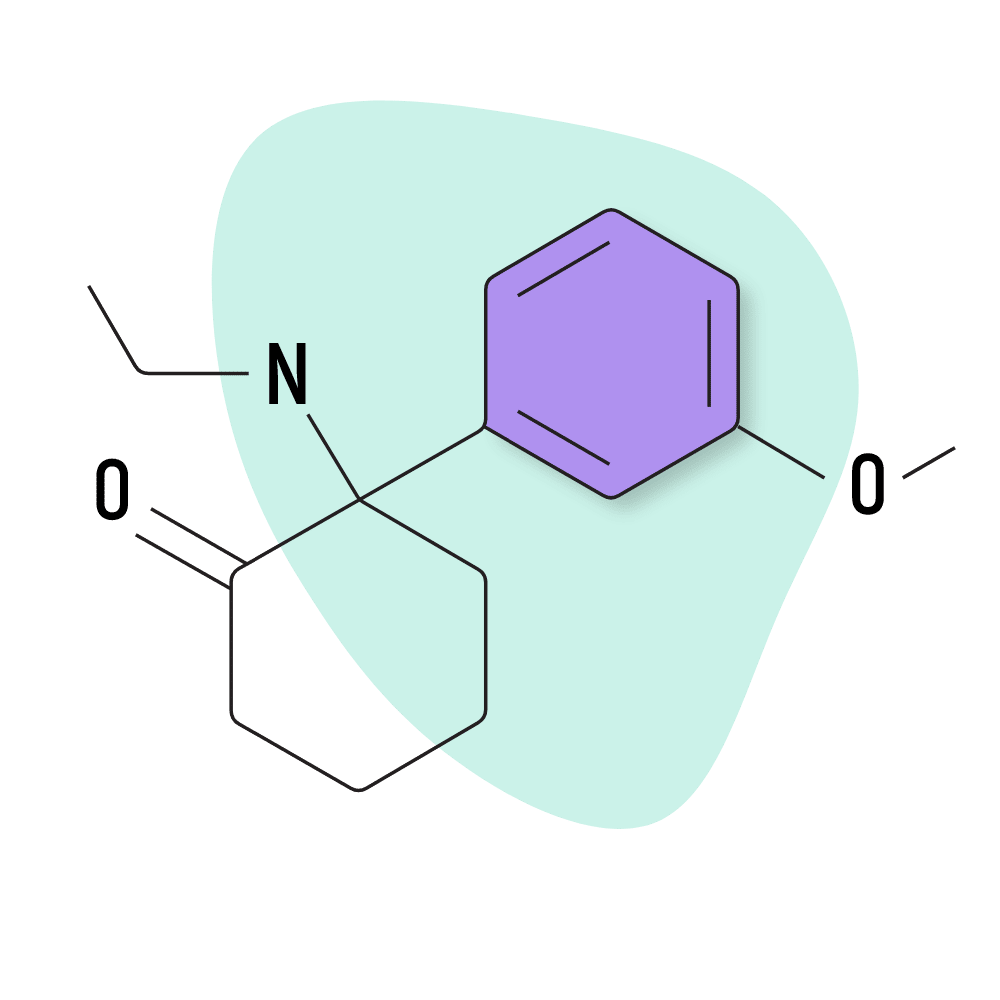
MXE (Methoxetamine)
MXE (AKA Mexxy) was allegedly created in an attempt to create a ketamine alternative that lacked the negative effects the drug has on the urinary tract. Because MXE is more potent than ketamine, smaller doses are needed to produce similar results — this would, in turn, reduce the levels of toxic metabolites that end up in the bladder. With that said, frequent users of MXE still report the same kinds of bladder disease as those found in frequent ketamine users.
The effects of MXE are very similar to ketamine — producing mild stimulation, euphoria, and feelings of disconnection from the body and environment, starting in low doses, and becoming more pronounced as the dose increases. High doses of MXE can result in K-hole experiences very similar to ketamine.
One marked difference between ketamine and MXE is that MXE appears to be less reliable at inducing a state of “euphoric nothingness,” in which the user exists in a place outside time and space, all alone but free from human emotion like grief or loneliness. Instead, MXE often produces experiences that are both terrifying and tragic — which is a leading theme in the many negative trip reports for this drug online.
While the effects of MXE are generally compared to ketamine, it is structurally more similar to eticyclidine (PCE) and 3-MeO-PCE.
There are also a few newer analogs of MXE, including MXPr and MXM — each of which has a similar effect profile. But varying levels of potency.
The receptor Ki values for MXE are reported at 259 nM for the NMDA receptor (antagonistic) and 481 for the serotonin transporter (inhibitor). It was found to have little to no affinity for the dopamine and norepinephrine transporters, as well as the σ1 and σ2 receptors.
MXE is one of the few arylcyclohexylamine analogs specifically included on the list of Schedule I restricted substances — most other compounds in this family are illegal by proxy, as per the Federal Analog Act.
MXE Specs:
| Chemical Name | Methoxetamine |
| Status | Research Chemical 🧪 |
| Duration of Effects | 3–6 hours |
| Estimated Threshold Dose | 5 mg |
| Common Dose | 10–50 mg |
| PubChem ID | 52911279 |
| CAS# | 1239943-76-0 |
IUPAC Name: (R/S)-2-(3-Methoxyphenyl)-2-(ethylamino)cyclohexanone
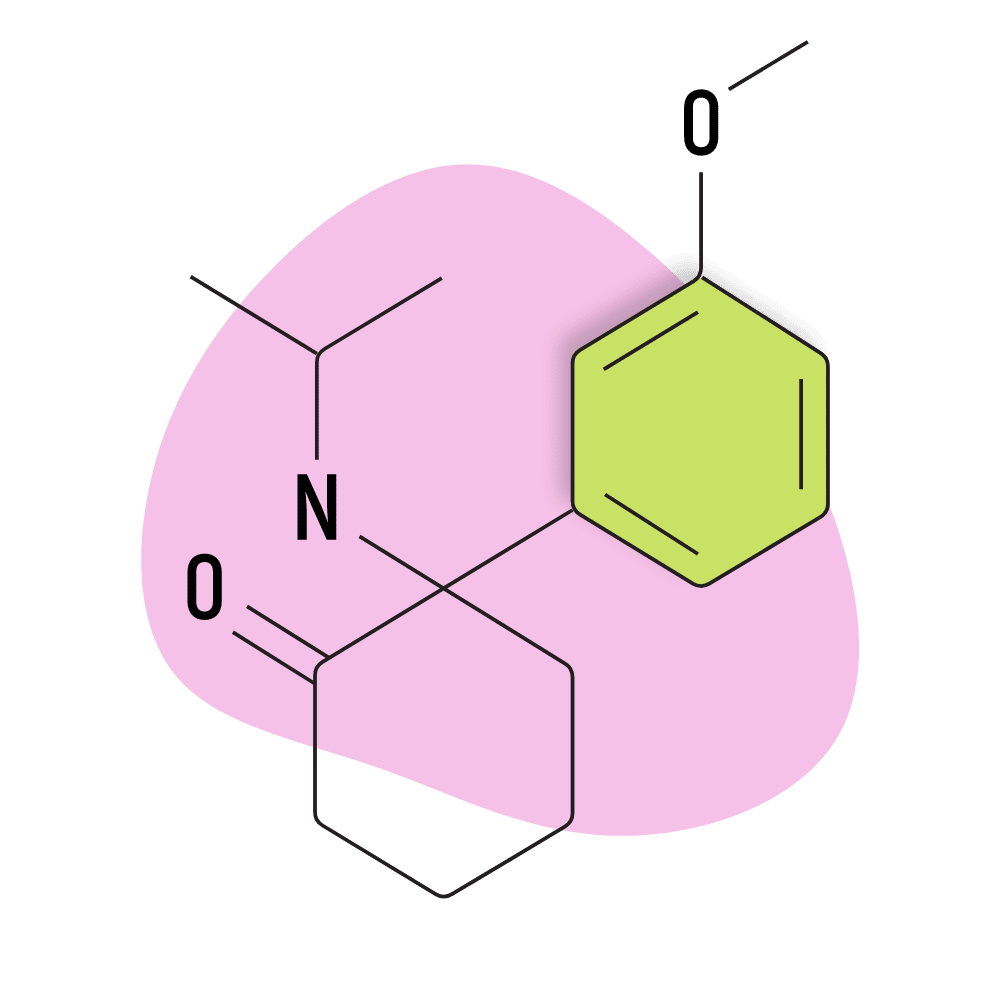
MXiPr (Methoxisopropamine)
MXiPr (AKA 3-MeO-2′-Oxo-PCiPr or methoxisopropamine) is a structural analog of MXE. This compound is relatively new to the market — with the first mention of the drug on designer drug markets only appearing in 2020.
Not much is known about the origin, but there are a lot of reports that the first and second batches had a strong chemical smell — first like plastic, and then like “Pokemon cards.” Many people have been hesitant to try this compound as a result of these claims — worrying that both batches contained potentially toxic impurities. This worry is present regardless of the way a particular compound smells. Impurities and contaminants in the research chemical space are rife.
The effects are suggested to be most similar to MXE but with a stronger sedative component, even in lower doses, and more visual (both eyes open and eyes closed).
The receptor Ki values for MXiPr have not yet been reported.
There are a variety of other compounds with the same PCiPr backbone (i-Propylamino), including 2’-Oxo-PCiPr, 3-Cl-PCiPr, 3-HO-PCiPr, 3-MeO-PCiPr, and 3-Me-PCiPr. None of these compounds have been explored in any meaningful way to date.
MXiPr Specs:
| Chemical Name | Methoxisopropamine |
| Status | Research Chemical 🧪 |
| Duration of Effects | 3–6 hours |
| Estimated Threshold Dose | 5 mg |
| Common Dose | 10–50 mg |
| PubChem ID | 155923150 |
| CAS# | Not Available |
IUPAC Name: 2-(isopropylamino)-2-(3-methoxyphenyl)cyclohexan-1-one
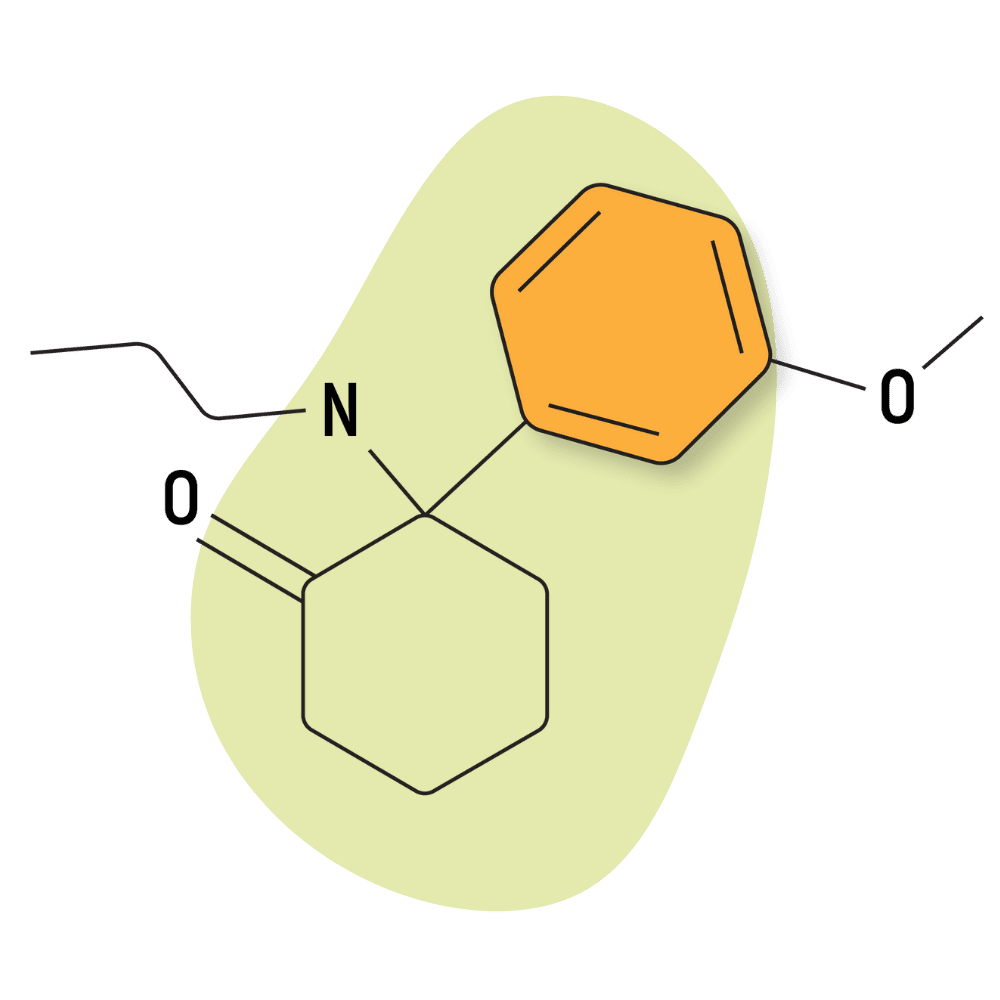
MXPr (Methoxpropamine)
MXPr hasn’t been around long, but it’s become quite popular over the past year. Many of its proponents suggest it’s especially colorful compared to other arylcyclohexylamines. It’s described as having a “warm” feeling, which is in opposition to the “cold” effects of drugs like ketamine.
Some people report this drug to be “empty” in terms of effects — but this appears to stem from its milder dissociative effects that make it harder to achieve a full K-hole experience. MXPr still produces feelings of dissociation and trippy closed and open-eye hallucinations, but without the loss of control and abysmal qualities associated with other members of the especially potent PCPr (n-Propylamino) group — see 3,4-MD-PCPr, 3-MeO-PCPr, 3-Me-PCPr, and PCPr.
Many of the compounds in this group have abnormally high selectivity for the NMDA receptors. There are no established Ki values for the binding capacity of MXPr on this receptor to accurately compare this drug with other members of the group — but anecdotal reports from people using the drug suggest it’s lower than usual. It’s likely this drug affects the other receptor sites (DAT, NET, SERT, sigma 1, and sigma 2 receptors) much differently than these related compounds.
MXPr Specs:
| Chemical Name | Methoxpropamine |
| Status | Research Chemical 🧪 |
| Duration of Effects | 1–2 hours |
| Estimated Threshold Dose | 5 mg |
| Common Dose | 10–60 mg |
| PubChem ID | 155817932 |
| CAS# | 2504100-71-2 |
IUPAC Name: 2-(3-Methoxyphenyl)-2-(propylamino)cyclohexanone
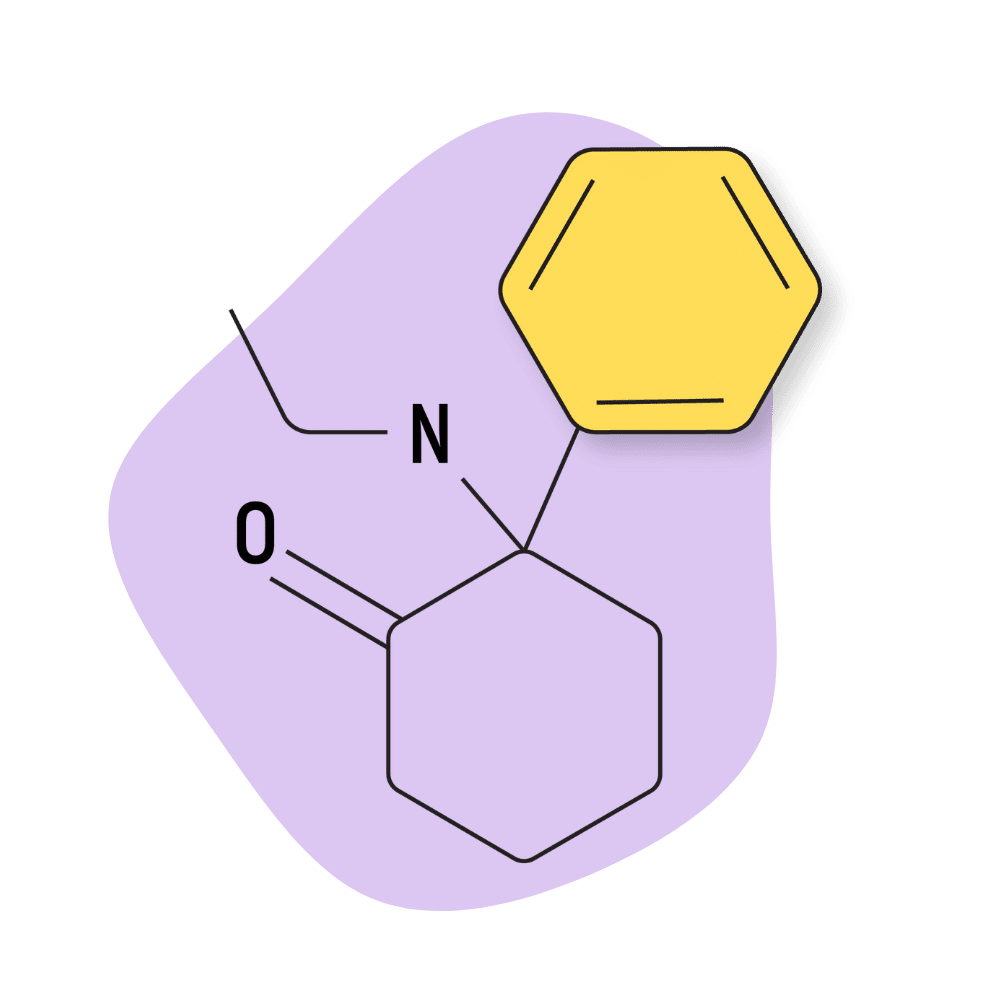
O-PCE (2-Oxo-PCE, Eticyclidone)
2-Oxo-PCE (AKA eticyclidone or O-PCE) is most similar to DCK — the only difference is that O-PCE has a one-carbon chain, while DCK has a 2-carbon chain in the same position. This difference is subtle but produces distinct changes in the effect profile.
Compared to DCK and ketamine, O-PCE is significantly more stimulating and has fewer inhibitory effects on the senses. This compound doesn’t normally exert its dissociative effects until 3 or 4 hours into the trip as it begins to wear off.
Because of the stimulating nature of this compound, O-PCE is often referred to as “chaotic” and “wonky,” — producing unexpected urges and bizarre behavior — especially in higher doses. In low to moderate doses, it’s usually described as warm and euphoric, with less of the heavy dissociative effects of other drugs like DCK or ketamine.
The receptor Ki values for O-PCE have not yet been reported.
O-PCE Specs:
| Chemical Name | N-ethyldeschloroketamine |
| Status | Research Chemical 🧪 |
| Duration of Effects | 3–6 hours |
| Estimated Threshold Dose | 1 mg |
| Common Dose | 3–15 mg (Oral & Insufflated) |
| PubChem ID | 132989542 |
| CAS# | 6740-82-5 |
IUPAC Name: 2-(ethylamino)-2-phenyl-cyclohexanone
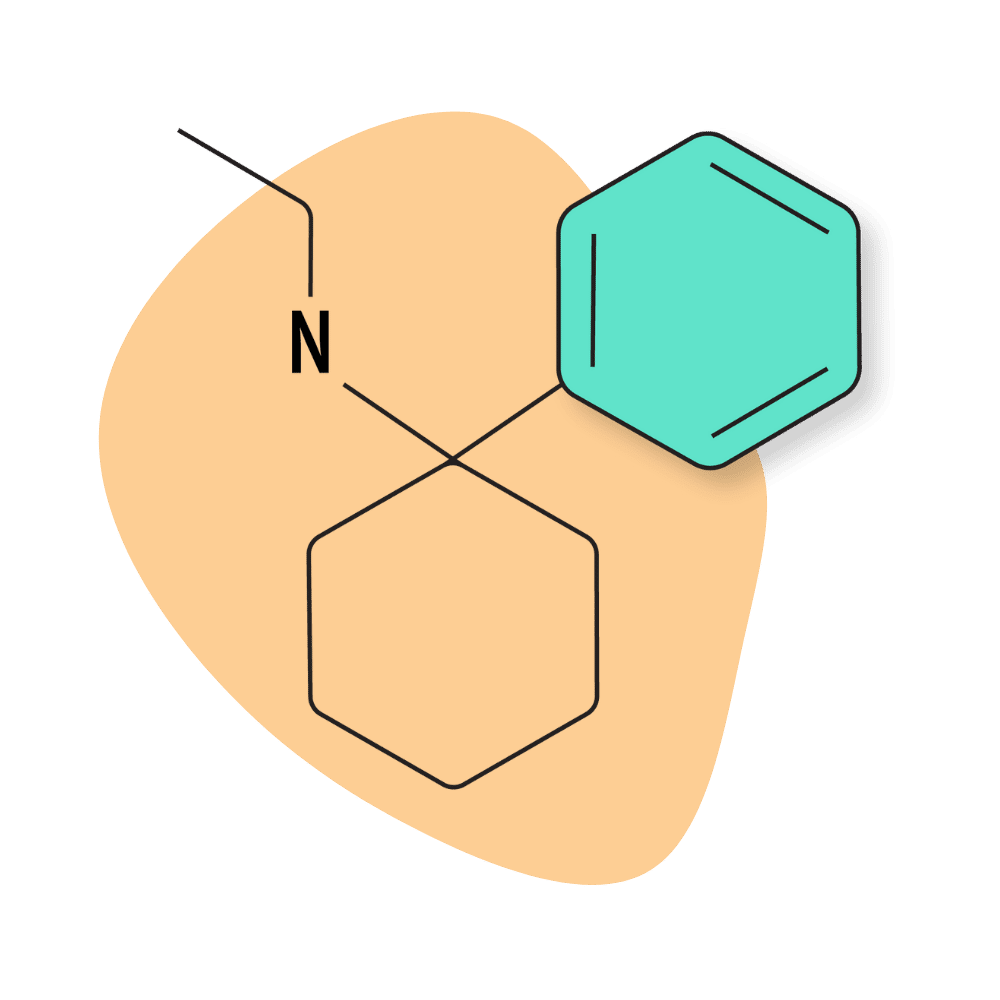
PCE (Eticyclidine)
PCE was the second arylcyclohexylamine Parke-Davis investigated during their early research on dissociative anesthetics. At the time, they referred to the chemical as CI-400.
PCE was almost developed as a replacement for PCP until they discovered ketamine which displayed much more ideal traits (less incidence of emergence delirium, short half-life, and reliable effects at various intravenous dosages). The company eventually abandoned the project to focus on ketamine.
Despite the project getting canned, PCE ended up on the street a couple of years later, where it became a drug of abuse in a similar manner to PCP. Effects-wise, PCE is most similar to PCP — but even stronger. It never quite became as popular as PCP because of its acrid taste and propensity for causing nausea in users at all doses. PCE analogs such as 3-MeO-PCE and O-PCE are more popular because they’re less likely to induce nausea and vomiting than their parent compound.
Other PCE analogs include 3,4-MD-PCE, 3-HO-PCE, 3-MeO-PCE, 3-Me-PCE, DMXE, FXE, HXE, MXE, O-PCE, and tiletamine. Most of the compounds in this group have Ki values for the NMDAR below 100 (suggesting high affinity). This potency makes PCE more difficult to use safely — just a tad too much can result in an entirely different experience than one might expect. K-holing on PCE has been reported at doses as low as 15 mg.
PCE results in the same issues with bladder damage as ketamine and other arylcyclohexylamines — however, the higher potency allows for much smaller doses to exhibit the same level of effects as something like ketamine and is therefore considered marginally safer in this respect but more dangerous in others (such as the formation of Olney’s lesions).
PCE Specs:
| Chemical Name | Eticyclidine |
| Status | Research Chemical 🧪 |
| Duration of Effects | 4–7 hours |
| Estimated Threshold Dose | 1 mg |
| Common Dose | 5–15 mg |
| PubChem ID | 16622 |
| CAS# | 2201-15-2 |
IUPAC Name: N-Ethyl-1-phenylcyclohexylamine
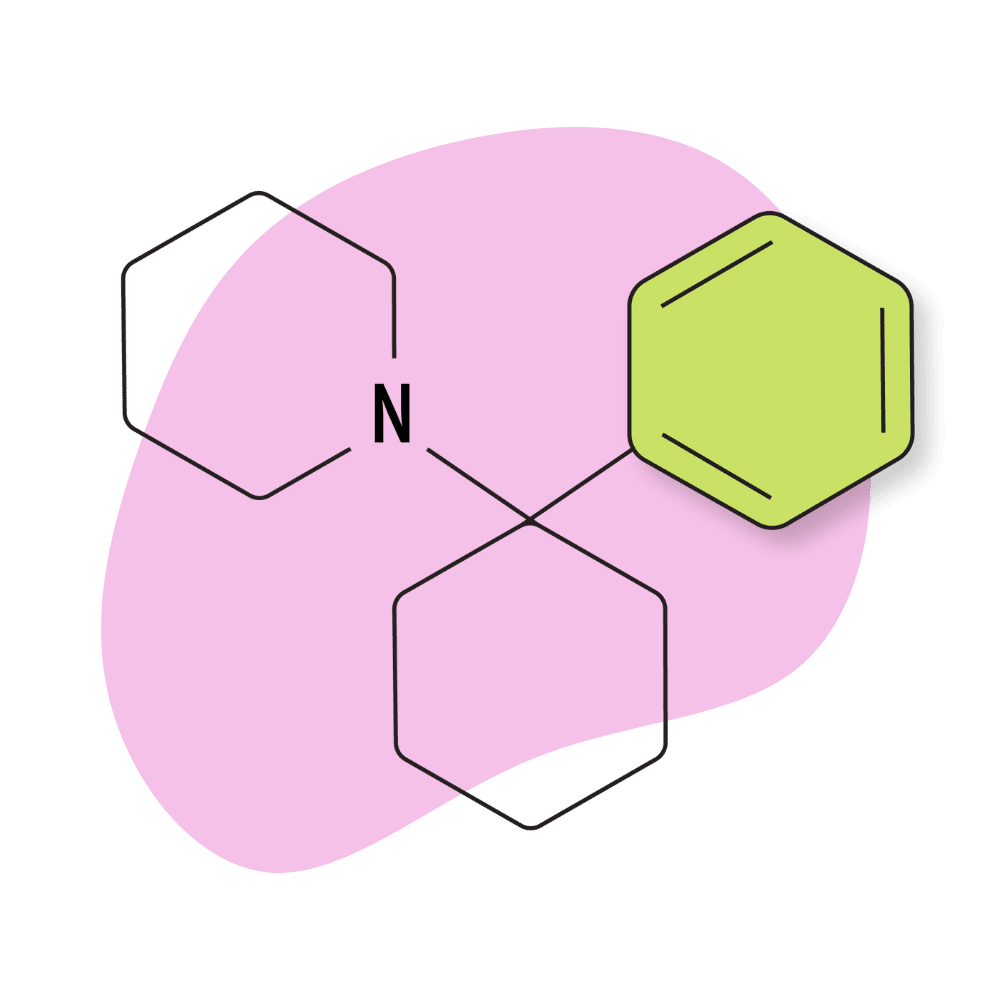
PCP (Phencyclidine)
Phencyclidine has had a long and checkered history. It started as an anesthetic drug used during surgeries (branded as Sernyl®) and ended up on the streets as a cheap drug of abuse. It became the target of anti-drug propaganda by the US federal government with stories that PCP could turn people into bulletproof, face-eating maniacs with just a single puff.
PCP was also at one time the most common deception drug — being sold as LSD, mescaline, psilocybin, cocaine, THC, and others. In the first half of the 1970s, less than 9% of all samples containing PCP were actually sold as PCP [21].
While the stories about PCP are certainly exaggerated, PCP isn’t without its risks. It’s well established to be an addictive and habit-forming substance with significant consequences resulting from long-term compulsive use.
PCP is widely used today as a recreational drug. It’s extremely cheap to produce, offers exceptionally strong hallucinogenic potency, and provides a long-lasting high.
Phencyclidine is often used as the baseline for comparing other arylcyclohexylamines. It provides feelings of stimulation, euphoria, and dissociation at lower doses and trippy out-of-body hallucinations in higher doses. Users often report feeling as though they’re floating or in some sort of dream-like state.
Many people experience what they believe to be cosmic insights about the nature of reality or the world around them while under the effects of PCP. It’s common for people to experience sensations that can’t be put into words — like watching themselves without needing a mirror, having their body teleported to far away places without their mind, or being unaffected by time.
PCP can also produce unwanted effects, such as feelings of aggression, disinhibition and increased risk-taking behavior, and confusion.
Despite the creation of many new arylcyclohexylamines over the years, PCP remains one of the most potent members ever discovered (but less potent than 3-MeO-PCPr, 3-MeO-PCPy, 3-HO-PCE, and 3,4-MD-PCPr).
The receptor Ki values for PCP are reported at 57.9 nM at the NMDA receptors (very potent), 430 for the dopamine transporters (moderate potency), 2,234 nM for the serotonin transporters (weak potency) — but negligible activity on the norepinephrine transporters.
PCP Specs:
| Chemical Name | Phencyclidine |
| Status | Discontinued |
| Duration of Effects | 4–8 hours |
| Estimated Threshold Dose | 1 mg |
| Common Dose | 2–15 mg |
| PubChem ID | 6468 |
| CAS# | 77-10-1 |
IUPAC Name: 1-(1-Phenylcyclohexyl)piperidine
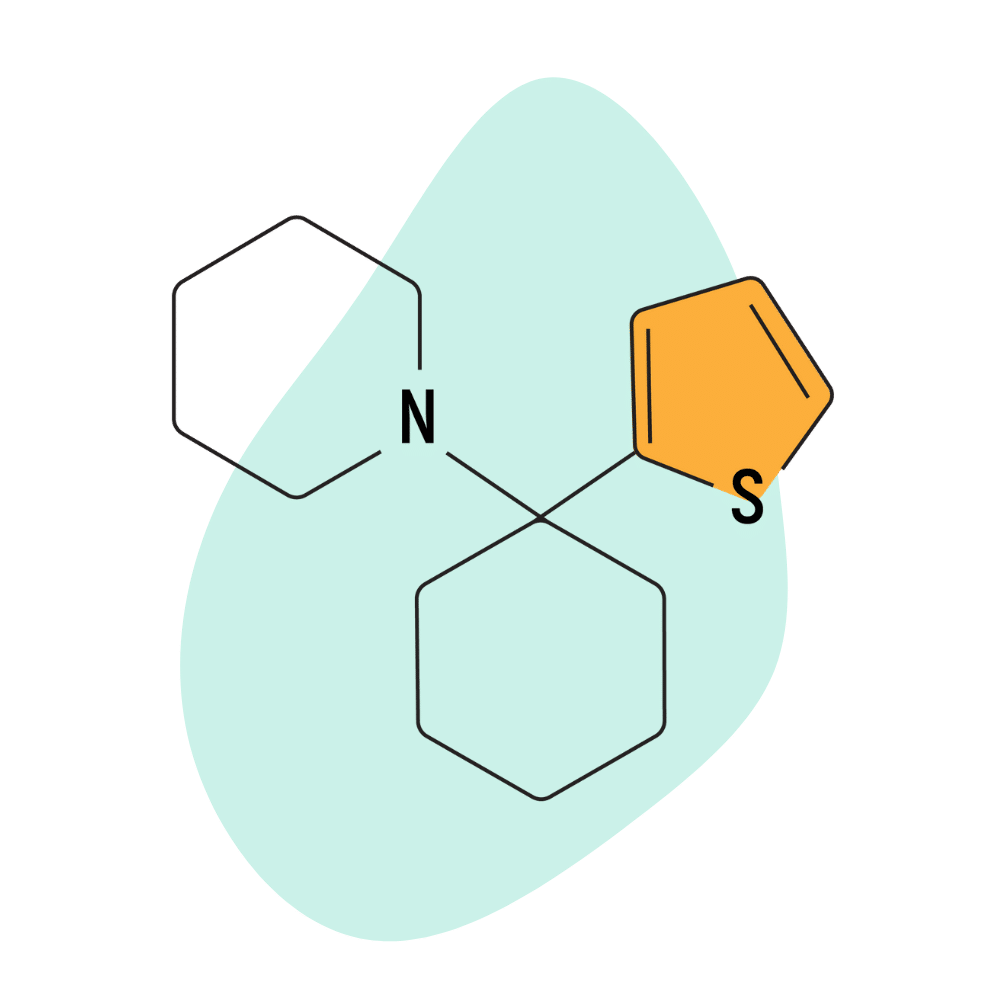
TCP (Tenocyclidine)
TCP was patented in the early 1960s by Parke-Davis — the same company to patent and eventually brought PCP and ketamine to market.
This compound is essentially a PCP molecule with a thiophene ring rather than a benzene ring. The rest of the chemical structure is identical between the two molecules. This alteration doesn’t make a massive amount of difference — in fact, Parke Davis considered it to be interchangeable with PCP aside from having a slightly higher potency than PCP by weight and a slightly longer duration of effects.
Parke-Davis focused on developing PCP as a medicine and shelved their research on TCP. After the compound ended up on the street, the US government eventually placed it on the Schedule I restricted drug list — marking it as the very first arylcyclohexylamine to earn the distinction. Regulators viewed it as a drug with a high potential for abuse, but since it wasn’t approved by the FDA, like PCP, it was without medical value.
The receptor binding (Ki) values for tenocyclidine have not been reported but are believed to share a similar profile to that of PCP.
Tenocyclidine Specs:
| Chemical Name | Tenocyclidine |
| Status | Research Chemical 🧪 |
| Duration of Effects | Unknown |
| Estimated Threshold Dose | 1 mg |
| Common Dose | Unknown |
| PubChem ID | 62751 |
| CAS# | 21500-98-1 |
IUPAC Name: 1-[1-(Thiophen-2-yl)cyclohexyl]piperidine (TCP)
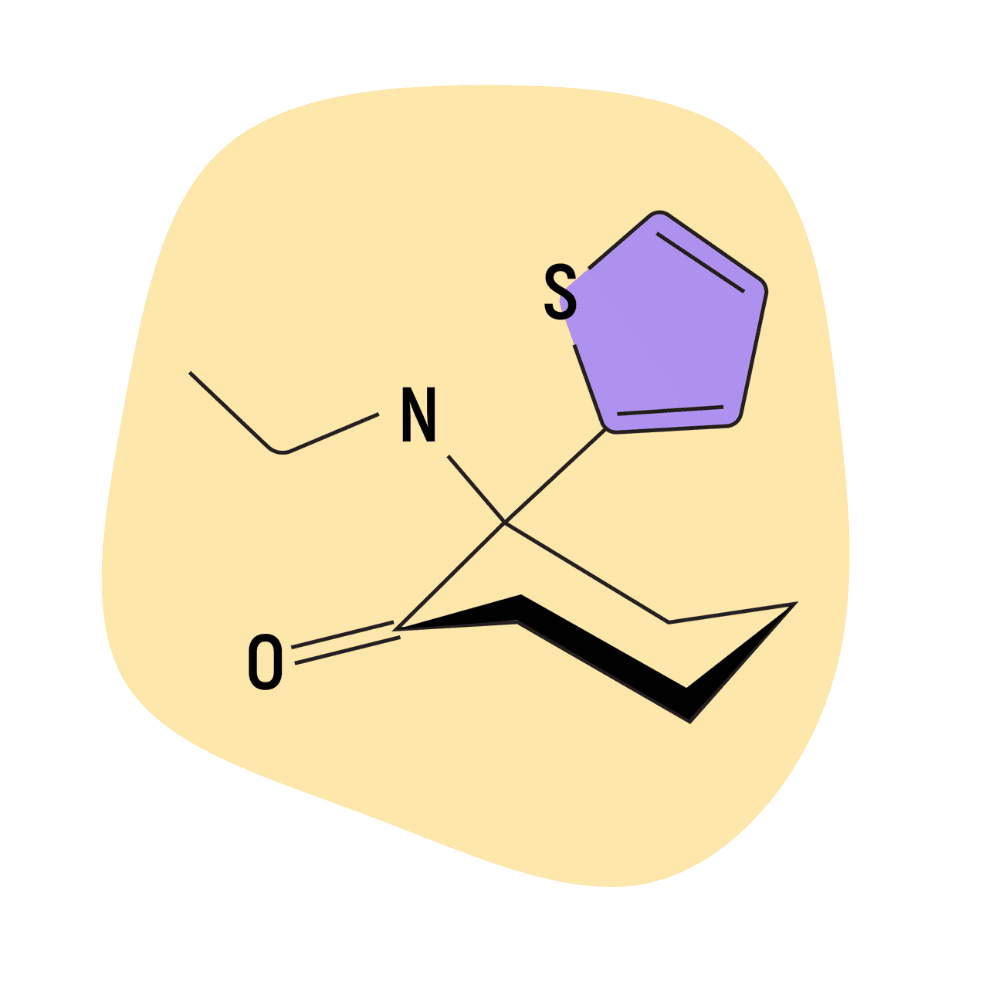
Tiletamine
Tiletamine (AKA CI-634) is a dissociative anesthetic primarily used in veterinary medicine. It’s primarily used in the form of a combination product called Telazol — which is a 50/50 mix of tiletamine and zolazepam.
There are reports of this drug being used recreationally, but the reviews are a bit of a mixed bag. Some users claim it’s highly underrated, capable of producing almost DMT-like visuals alongside the conventional dissociative experience.
Others suggest tiletamine provides only a mild dissociative high followed by 24–48 hours of nausea and flu-like symptoms.
Tiletamine is very expensive compared to other dissociatives but surprisingly common on the market. This is likely because it’s stolen from vet clinics and sold on the street.
The problem is that tiletamine stolen from vet clinics often comes pre-mixed with benzos (primarily zolazepam), which adds a strong sedative component that effectively inhibits the desired dissociative effects.
Tiletamine Specs:
| Chemical Name | Tiletamine |
| Status | Approved For Veterinary Use |
| Duration of Effects | 3–6 hours |
| Estimated Threshold Dose | 3 mg |
| Common Dose | 5–30 mg |
| PubChem ID | 26533 |
| CAS# | 14176-49-9 |
IUPAC Name: 2-Ethylamino-2-(2-thienyl)cyclohexanone
How Do Arylcyclohexylamines Work?
Arylcyclohexylamines interfere with the function of glutamate — a key neurotransmitter involved with brain function (and, therefore, consciousness). This results in a powerful dissociation of the mind from the body — all without extinguishing consciousness.
Most of the arylcyclohexylamine class of drugs work by binding to the PCP binding site of the NMDA receptor [8]. PCP was the first compound ever studied and was instrumental in understanding the NMDA receptors. However, most members of this group — not just PCP — bind to this same receptor.
Once bound, arylcyclohexylamines block the effects of NMDA, resulting in their characteristic dissociative effects.
Arylcyclohexylamines also interact with the dopamine transporter (DAT), norepinephrine transporter (NET), and serotonin transporter (SERT) to exert additional effects on mood and cognition.
For example, PCP is a strong stimulant in addition to its dissociative qualities. This effect stems from the compound’s affinity for dopamine, norepinephrine, and serotonin transporters. By blocking the reuptake of these neurotransmitters, these compounds exert a potent amphetamine-like effect — boosting cognition, increasing vigilance and alertness, and raising body temperature.
One study found PCP was 8 times more potent than D-amphetamine and 34 times more potent than methylphenidate at inhibiting serotonin (5-HT) uptake [12].
PCP has also been found to interact with the sigma receptors — especially the sigma 2 receptor — which are involved with a wide range of neurological functions. The specific function of these receptors is not well understood. Several psychoactive drugs have been found to interact with these receptors in addition to PCP and PCP-analogs, including cocaine, morphine, fluvoxamine, methamphetamine, and DXM (dextromethorphan).
The binding affinity for each arylcyclohexylamine varies — some have a stronger dopamine component; others are stronger NMDA antagonists. These changes in binding affinity impact the drugs’ subjective effect profile — making them stronger dissociatives, sedatives, or stimulants than other members of the group.
The arylcyclohexylamine class is also notoriously biphasic in terms of their effects — this means they produce different effects in low doses than they do in higher doses. In general, lower doses are stimulating, while higher doses are sedative and cataleptic. Most members of this group share these qualities.
List of Primary Target Receptors for the Arylcyclohexylamine Class:
| Receptor | Interaction | Effect Outcome |
| N-Methyl-D-Aspartate (NMDA) Receptors | Antagonistic (Inhibits) | Dissociation, sedation, neuroprotective (dose-dependant), |
| Opioid Receptors | Agonistic (Stimulates) | Analgesia, sedation, euphoria, anxiolysis |
| Dopamine (D2) Receptors | Agonistic (Stimulates) | Hallucinations, euphoria, mental & physical stimulation, thermogenesis |
| Dopamine Transporters (DAT) | Antagonist (Inhibits) | Stimulation, hallucinations, euphoria, thermogenesis |
| Norepinephrine Transporters (NET) | Antagonist (Inhibits) | Stimulation, thermogenesis |
| Serotonin Transporters (SERT) | Antagonist (Inhibits) | Euphoria, hallucinations |
| Sigma Receptors | Agonistic (Stimulates) | Hallucinations, dissociation |
| Nicotinic Acetylcholine (nACh) Receptors | Antagonistic (Inhibits) | Muscle-relaxation, intoxication |
N-Methyl-D-Aspartate (NMDA) Receptors
The NMDA receptors play a key role in synaptic plasticity, memory, and pain perception [42, 43]. These receptors are activated by glutamate, which is the primary excitatory neurotransmitter in the brain.
A variety of psychoactive drugs rely on an interaction with the NMDA receptors to exert their effects, including alcohol, GHB, xenon gas, nitrous oxide, methadone, and all the psychoactive arylcyclohexylamines. Blocking these receptors or otherwise inhibiting their function leads to a state of dissociation and inhibits the formation of memories. It also results in the characteristic dissociation and depersonalization inherent to this class — but the specific mechanism for how this works is still not well understood.
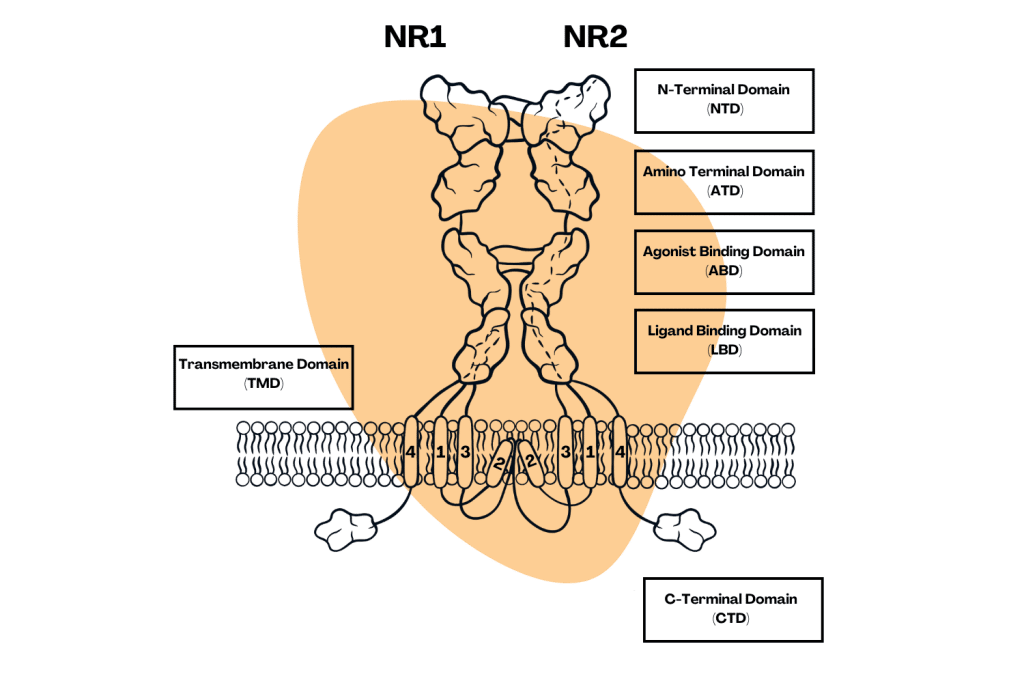
Strong inhibition of the NMDA receptors is responsible for the “K-hole” experience from ketamine and its related analogs, as well as the disinhibition and amnesia produced by drugs like alcohol and GHB.
The NMDA receptors are integral to human cognition. Dysfunction of these receptors is involved with a variety of disease states as well, including Alzheimer’s disease [44], Huntington’s disease [45], Epilepsy [46], depression [47], and tinnitus [48].
Opioid Receptors (Mu, Delta, Kappa)
The opioid receptors are a group of G protein-coupled receptors that mediate the body’s response to hormones and neurotransmitters and are involved in sensory perception (taste, touch, vision, and olfaction).
These receptors are intimately involved in both the sensation of pain and pleasure. There are three different types (mu- delta-, and kappa-opioid receptors). All three opioid receptor subtypes inhibit the transmission of pain when activated, but the psychoactive effects of each are unique due to differences in the way they’re involved with stress and hedonics (motivation via pleasure-reward pathway) [49].
The mu-opioid receptors (MOR) are largely responsible for the painkilling and antidepressant effects of certain arylcyclohexylamines, such as PCP and ketamine [50, 51]. These receptors are strongly involved in hedonics — producing feelings of euphoria and mediating positive reinforcement [52]. It’s likely this receptor plays a pivotal role in the addictive potential of the arylcyclohexylamine family.
The delta opioid receptors (DOR) have the strongest impact on emotional regulation and mood of the three opioid receptor types [53]. Activation of this receptor is linked with anxiolytic effects and positive affect [49]. Compounds with a strong affinity for delta-opioid activation are currently being investigated as future antidepressant medications.
The kappa opioid receptors (KOR) have the lowest involvement in hedonics and mood but are involved in stress response [54]. Agonism of this receptor subtype leads to feelings of dysphoria (sense of uneasiness), stress-like responses, and negative affect [49].


Salvinorin A — the active psychedelic compound in Salvia divinorum — has a high affinity for the kappa-opioid receptor [55]. It’s believed this effect is responsible for the bizarre hallucinations this plant produces when smoked. It’s believed (though not proven) that various arylcyclohexylamines owe some of their peculiar psychedelic actions to the kappa opioid receptor as well.
The effects on the opioid receptors of arylcyclohexylamines likely play a key role in the euphoriant, calming, and analgesic effects of these substances.
Dopamine (D1 & D2) Receptors
The dopamine receptors are a group of G-protein coupled receptors involved in the regulation of hormone production, hedonics (pleasure and reward pathway), and movement [56]. They’re the primary target for stimulant drugs like amphetamines, cocaine, and nicotine.
Many common antipsychotic agents used to treat neuropsychiatric disorders work through these receptors — such as haloperidol (blocks D2 to treat schizophrenia) or apomorphine (stimulates D2 to treat Parkinson’s disease).

There are five main types of dopamine receptors, but the most relevant in terms of the arylcyclohexylamines are D1 and D2. Both have similar functions but are distributed differently throughout the brain [57].
D1 receptors are primarily found in the striatum, which is a region of the brain involved in movement and motivation.
D2 receptors, on the other hand, are more diverse and can be found in a variety of different brain regions, including the striatum, the hippocampus, and the basal ganglia.
Several members of the arylcyclohexylamine group have been found to act as dopamine agonists, primarily at the D2 receptor subtype. It’s believed this interaction plays a key role in these drugs’ euphoric, stimulant, and addictive qualities.
Dopamine Transporters (DAT)
The dopamine transporters are special proteins embedded in the membranes of nerve cells responsible for the reuptake of dopamine into the synapses. Once reabsorbed, dopamine is either stored to be used again or broken down.
Dopamine transporters play a key role in the regulation of dopamine function and healthy neurological function.
Blocking this transporter causes dopamine to accumulate in the synapses, causing increased feelings of pleasure and energy [58].
Many psychoactive drugs with stimulant-like effects work by blocking the reuptake of dopamine — including many arylcyclohexylamines, cocaine, and various amphetamines. The arylcyclohexylamines with the strongest impact on DAT include PCP, 3-Me-PCPr, and 3-MeO-PCPy. All three of these compounds are notoriously stimulating.
Norepinephrine Transporters (NET)
Norepinephrine is a neurotransmitter and hormone. When produced by the adrenals, it’s used as a regulator for the fight or flight response, and when produced by neurons in the brain, it’s used to increase wakefulness and alertness, mood, and behavior [59, 60].
The norepinephrine transporters are responsible for reabsorbing spent norepinephrine back into the cell that released them [61]. They work in a similar capacity to the dopamine transporters and serotonin transporters but are specific for norepinephrine.
Drugs that inhibit the NETs, including arylcyclohexylamines, tend to have a stimulating action. Compounds like 3-MeO-PCPy, 3-Me-PCPr, and 3-Me-PCE have the strongest impact on NET.
Serotonin Transporters (SERT)
Serotonin is involved in the regulation of mood, behavior, appetite, sleep, pain perception, aggression, and much more [62].
The serotonin transporters are responsible for regulating serotonin levels by removing it from the synapses when it’s no longer needed [63]. They’re a subgroup of the SLC6 (solute carrier family 6) transporters which also includes DAT and NET as well as glycine transporters (GlyTs). Triple-reuptake inhibitors refer to any compound that inhibits the function of all SLC6 transporters. Many arylcyclohexylamine compounds are triple-reuptake inhibitors.
Blocking the SERT causes an elevation of serotonin in the synapses beyond normal levels. Therapeutic targets designed to block the serotonin transporter (such as SSRIs and SNRIs) are mainstay treatment options for depression [64].
Sigma Receptors
The sigma receptors (σ receptors) are a family of proteins in the brain. They’re a subtype of the classical opioid receptors but have a distinct role and function.
These receptors are believed to play a role in the regulation of pain, anxiety, and addiction [65] — but the specific function of these receptors is still not well understood.
There are two types of sigma receptors — sigma 1 (σ1) and sigma 2 (σ2). Both subtypes share similarities but have distinct roles. Many arylcyclohexylamines bind to both receptor types, and both are believed to play a key role in their qualitative effects.

Arylcyclohexylamines like PCP and ketamine, as well as other psychoactive drugs, including morphine, cocaine, fluvoxamine, methamphetamine, and DXM, were all found to interact with these receptors. It’s believed this mechanism contributes to the hallucinogenic, euphoriant, and stimulant effects of these drugs. It’s also involved in the psychomimetic effects and increased body temperature experienced with higher doses.
Nicotinic Acetylcholine (nACh) Receptors
The nACh belongs to the “cys-loop” superfamily of ligand-gated ion channels that also include GABA-A, glycine, and serotonin (5-HT3) receptors [28].
These receptors are broadly distributed around the central nervous system, which means they have a very wide range of functions. The nACh receptors are involved with the mediation of fast neurotransmission in the central and peripheral nervous systems [29]. These receptors are involved with various cognitive functions as well as synaptic transmission from nerve to muscle.

nACh receptors are involved in various neurodegenerative disorders, including Alzheimer’s disease [30, 32], schizophrenia [31], Parkinson’s disease [33], and depression [34]. They’re also believed to play a role in the addictive potential of nicotine [35] and could be involved in the addictive potential of the arylcyclohexylamine class as well.
There are a variety of natural toxins that block these receptors, including cone snail toxins (α-Conotoxins) [36], plant toxins (methyllycaconitine from aconitum, d-tubocurarine from Chondrodendron tomentosium, and strychnine from Strychnos nux vomica) [37,38,39], and snake toxins (α-cobratoxin and α-bungarotoxin) [40].
Arylcyclohexylamines were found to block these receptors, which may contribute to the muscle-relaxant, intoxicating, and toxic effect profiles of these substances.
Arylcyclohexylamine Receptor Binding Affinities (Ki & IC50 Values):
| Name | NMDAR Affinity | DAT Affinity | NET Affinity | SERT Affinity |
| 2-MeO-PCMo | Low ↑ 1,578 nM [6] (Ki) | Negligible >10,000 nM [6] (IC50) | Negligible >10,000 nM [6] (IC50) | Moderate ↑↑ 507 nM [6] (Ki) |
| 3′-MeO-PCPr | High ↑↑↑ 17.9 nM [5] (Ki) | Low ↑ 1,342 nM [5] (Ki) | Low ↑ 1,342 nM [5] (Ki) | Moderate ↑↑ 700 nM [5] (Ki) |
| 3′-MeO-PCPy | High ↑↑↑ 22.3 nM [5] (Ki) | High ↑↑↑ 96 nM [5] (Ki) | High ↑↑↑ 96 nM [5] (Ki) | Low ↑ 11 nM [5] (Ki) |
| 3′-HO-PCE | High ↑↑↑ 23 nM [Unverified] (IC50) | Unknown | Unknown | Unknown |
| 3′,4′-MD-PCPr | High ↑↑↑ 24.9 nM [5] (Ki) | Negligible >10,000 nM [5] (IC50) | Negligible >10,000 nM [5] (IC50) | Negligible >10,000 nM [5] (IC50) |
| 3′-HO-PCP | High ↑↑↑ 30 nM [9] (Ki) | Negligible >10,000 nM [5] (IC50) | Negligible >10,000 nM [5] (IC50) | Negligible >10,000 nM [6] (IC50) |
| 3′-MeO-PCE | High ↑↑↑ 30.4 nM [5] (Ki) | Low ↑ 1,528 nM [5] (Ki) | Low ↑ 1,528 nM [5] (Ki) | High ↑↑↑ 136 nM [5] (Ki) |
| 3′,4′-MD-PCE | High ↑↑↑ 35.5 nM [5] (Ki) | Negligible >10,000 nM [5] (IC50) | Negligible >10,000 nM [5] (IC50) | Moderate ↑↑ 637 nM [5] (Ki) |
| 3′-MeO-PCP | High ↑↑↑ 38.1 nM [5] (Ki) | Negligible >10,000 nM [5] (Ki) | Low ↑ 1,808 nM [5] (Ki) | Low ↑ 1,571 nM [5] (Ki) |
| 3′-Me-PCE | High ↑↑↑ 43.1 nM [5] (Ki) | Negligible >10,000 nM [5] (IC50) | Moderate ↑↑ 598 nM [5] (Ki) | High ↑↑↑ 117 nM [5] (Ki) |
| 3′-Me-PCPr | High ↑↑↑ 53.5 nM [5] (Ki) | Moderate ↑↑ 113 nM [5] (Ki) | Moderate ↑↑ 264 nM [5] (Ki) | Moderate ↑↑ 853 nM [5] (Ki) |
| PCP (Phencyclidine) | High ↑↑↑ 57.9 nM [5] (Ki) | Moderate ↑↑ 430 nM [7] (Ki) | Negligible >10,000 nM [11] (IC50) | Low ↑ 2,234 nM [11] (Ki) |
| 3′,4′-MD-PCP | High ↑↑↑ 62.8 nM [8] (Ki) | Unknown | Unknown | Unknown |
| 3′-MeO-PCMe | High ↑↑↑ 145.1 nM [5] (Ki) | Low ↑ 3,238 nM [5] (Ki) | Low ↑ 3,238 nM [5] (Ki) | Low ↑ 1,368 nM [5] (Ki) |
| 3′-Me-PCMo | Moderate ↑↑ 201.8 nM [6] (Ki) | Negligible >10,000 nM [6] (IC50) | Negligible >10,000 nM [6] (IC50) | Moderate ↑↑ 412 nM [6] (Ki) |
| 3′-MeO-PCMo | Moderate ↑↑ 252.9 nM [6] (Ki) | Negligible >10,000 nM [6] (IC50) | Negligible >10,000 nM [6] (IC50) | Moderate ↑↑ 697 nM [6] (Ki) |
| MXE (Methoxetamine) | Moderate ↑↑ 259 [11] (Ki) | Negligible >10,000 nM [11] (IC50) | Negligible >10,000 nM [11] (IC50) | Moderate ↑↑ 481 [11] (Ki) |
| Ketamine | Moderate ↑↑ 323.9 nM [6,10] (Ki) | Negligible >10,000 nM [11] (IC50) | Negligible >10,000 nM [11] (IC50) | Negligible >10,000 nM [11] (IC50) |
| PcMo | Moderate ↑↑ 334.1 nM [6] (Ki) | Negligible >10,000 nM [6] (IC50) | Negligible >10,000 nM [6] (IC50) | Negligible >10,000 nM [6] (IC50) |
| 3′,4′-MD-PCMo | Moderate ↑↑ 425.5 nM [6] (Ki) | Negligible >10,000 nM [6] (IC50) | Negligible >10,000 nM [6] (IC50) | Negligible >10,000 nM [6] (IC50) |
| 4′-MeO-PCP (Methoxydine) | Moderate ↑↑ 620 nM [5] (Ki) | Low ↑ 1,811 nM [6] (Ki) | Low ↑ 1,811 nM [5] (Ki) | Moderate ↑↑ 900 nM [5] (Ki) |
| 4′-MeO-PCMo | Low ↑ 2118 nM [6] (Ki) | Negligible >10,000 nM [6] (IC50) | Negligible >10,000 nM [6] (IC50) | Negligible >10,000 nM [6] (IC50) |
Ki vs. IC50 values:
There are two main ways researchers measure the affinity of a given molecule for various receptors — Ki and IC50.
Ki describes the amount of a substance needed to reach 50% saturation of the target receptors, while IC50 refers to the amount of an inhibitor needed to achieve 50% inhibition of the receptor.
In simple terms, Ki refers to the binding affinity of a compound, while IC50 refers to its functional strength.
Both measure the same thing but from different angles.
Most companies conducting drug research prefer using the Ki metric because IC50 doesn’t take into account factors like binding equilibrium.
Regardless, the most important thing is to use the same scale (Ki or IC50) when comparing a compound’s binding affinity. Unfortunately, different researchers may prefer one over the other, so it’s common to see both metrics used.
Generally speaking, the lower the number, the stronger a compound’s binding affinity. This is because it requires less of the compound to inhibit the activity of that receptor.
Frequently Asked Questions
1. What’s the Strongest Arylcyclohexylamine Psychedelic?
The strongest arylcyclohexylamine in NMDA-binding affinity is 3-MeO-PCPr with a Ki value of 17.9 nM, followed by 3-MeO-PCPy, 3-HO-PCE, 3,4-MD-PCPr, and 3-HO-PCP.
This drug class is not well studied, and there are only a small handful of unofficial trip reports available for the vast majority of these substances. This makes it difficult to definitively rank the potency of this class. We believe the best way to make an educated guess is to compare the NMDA activity.
However, other variables play a role as well, such as bioavailability, SERT, DAT, NET, sigma-1 and sigma-2, and nACHr binding activity. All of these receptors collaborate to produce the resulting effects of these drugs, so this list is not definitive.
We will update this list as more information becomes available.
Estimated ranking of arylcyclohexylamines in order of potency:
- 2-MeO-PCMo
- 3′-MeO-PCPr
- 3′-MeO-PCPy
- 3′-HO-PCE
- 3′,4′-MD-PCPr
- 3′-HO-PCP
- 3′-MeO-PCE
- 3′,4′-MD-PCE
- 3′-MeO-PCP
- 3′-Me-PCE
- 3′-Me-PCPr
- PCP (Phencyclidine)
- 3′,4′-MD-PCP
- 3′-MeO-PCMe
- 3′-Me-PCMo
- 3′-MeO-PCMo
- MXE (Methoxetamine)
- Ketamine
- PcMo
- 3′,4′-MD-PCMo
2. Do Dissociatives Cause Brain Damage?
Excessive use of NMDA antagonists (including arylcyclohexylamines) have been known to cause a specific type of brain damage called Olney’s lesions — which refer to clusters of dead neurons in the white matter of the brain. This type of brain damage results in problems with memory loss, difficulty focusing, and various other cognitive deficits.
This effect has only been reported with usage patterns that can be described as abuse (very high doses or long-term, repetitive use).
Low doses of ketamine and other arylcyclohexylamines appear to have the opposite effect and are thought to protect the neurons from damage or even promote recovery.
3. Are Arylcyclohexylamines Toxic?
All arylcyclohexylamines are toxic in very high doses, but only a few appear to be toxic in low doses. Repetitive or compulsive use of these drugs, even in low doses, also appears to have toxic side effects — especially on the brain and bladder.
The effects of overdose on the arylcyclohexylamine class vary depending on the substance — but many run the risk of causing hyperexcitability, increased body temperature, delusions, disinhibition, seizures, coma, and death.
Lower-dose arylcyclohexylamines carry the biggest risk of causing bladder toxicity.
The best way to measure the toxic dose of a substance is to look at the LD50 — which refers to the lethal dose required to kill 50% of the population tested (usually rodents).
The LD50 level has not been assessed for the vast majority of arylcyclohexylamines, but the high-potency PCP and the low-potency ketamine have been tested and give us a baseline for this class.
The LD50 for PCP has been reported to be around 0.02854 mg/kg (IP) [13] and 11.3 mg/kg (IV) for ketamine [41].
4. How Do You Pronounce Arylcyclohexylamine?
Arylcyclohexylamine is pronounced as “air-il-sigh-kloh-hex-ih-luh-meen.”
5. What Do Dissociatives Feel Like?
Dissociatives cause users to feel like they’ve been disconnected from their bodies and/or the environment around them. They’re sometimes teleported to a different location, or may feel like they’re sitting somewhere outside their body — either watching themselves or simply aware that their body is no longer present.
Most users find it difficult to describe the dissociative experience following a trip because it often requires the use of nonsensical language like “I visited a place outside of time” or “everything was close and far at the same time.”
Of course, none of this makes sense using verbal language, but the dissociative experience can very easily cause these kinds of paradoxical or nonsensical sensations.
The dissociative state manifests differently for everybody, but some common descriptions of this state include the following:
- Feeling like you’re watching yourself from somewhere else
- Feeling like you’re outside your body — sometimes control of the body is maintained, other times the body appears to act on autopilot
- Time doesn’t make sense — something that felt like hours could have taken place in a matter of seconds, and things that felt like a few seconds could have spanned several hours
- Very strong closed and open-eye hallucinations may take place, especially with higher doses — the depths of the dissociative state are some of the most profoundly psychedelic experiences known
- You may not recognize yourself in the mirror (frequent users often recommend avoiding looking in the mirror on dissociatives because this effect can be unsettling)
- You may experience glimpses of past experiences flashing into your field of vision like a slideshow reel
- You may remain grounded somewhat in reality or be removed from it entirely
6. What is the Mechanism of Action For Dissociatives?
The primary mechanism of action for dissociative drugs is a blockage of the NMDA receptors. This prevents glutamate from binding to these receptors resulting in widespread interruptions in neurotransmission in the higher regions of the brain.
Other factors involved include the activation of the kappa-, mu-, and delta-opioid receptors, activation of the dopaminergic receptors, activation of the sigma-1 and sigma-2 receptors, and inhibition of the dopamine, serotonin, and norepinephrine transporters.
8. What Other Drugs Act As Dissociatives?
Other drugs with dissociative effects include nitrous oxide and xenon gas, MK-801, and dextromethorphan (DXM).
Alcohol, gamma-hydroxybutyrate (GHB), and some benzodiazepines can also exert a dissociative-like effect — but these drugs are nowhere near as strong as the others mentioned above.
9. Which Arylcyclohexylamines Can Be Vaped?
Most arylcyclohexylamines can be vaped or smoked. In fact, this is one of the primary modes of administration people use for taking PCP.
There is no safety information on vaporizing arylcyclohexylamines, so it’s unclear what the short or long-term health consequences may be.
One salient danger of this method of administration is that any heat-stable members of this group will likely be much stronger when smoked — and kick in much quicker. This could make overdosing much more likely if people are not very careful with their consumption.
Other substances may not be heat stable and will have little to no effect when vaped. More information is needed.
10. When Was the First Arylcyclohexylamine Invented?
The first arylcyclohexylamine ever reported is a compound called 1-(1-Phenylcyclohexyl)amine (PCA), which was reported back in 1907. This compound is the precursor to PCP and many other members of the group.
Nearly half a century later, the second arylcyclohexylamine, N-ethyl-1-phenylcyclohexylamine (PCE), was discovered in 1953. This kicked off a chain reaction of research on this group. The following year 1-(1-Phenylcyclohexyl)morpholine (PCMo) was included in a German patent.
Two years later, in 1956, PCP was discovered by Victor Maddox while working for Parke-Davis. This was the first arylcyclohexylamine to make it to market (approved in 1957).
A few years later, in 1962, Calvin L. Stevens (also working for Parke Davis as a consultant) discovered 2-oxo-PCE in 1962. Stevens’ lab later developed ketamine, which went on to become the most successful arylcyclohexylamine produced by Parke Davis.
Learn More: Videos & Other Sources
Hamilton Morris & Jason Wallach Lecture

References
- Stiller, R. L., Dayton, P. G., Perel, J. M., & Hug Jr, C. C. (1982). Gas chromatographic analysis of ketamine and norketamine in plasma and urine: nitrogen-sensitive detection. Journal of Chromatography B: Biomedical Sciences and Applications, 232(2), 305-314.
- Rosas, M. E. R., Patel, S., & Wainer, I. W. (2003). Determination of the enantiomers of ketamine and norketamine in human plasma by enantioselective liquid chromatography–mass spectrometry. Journal of Chromatography B, 794(1), 99-108.
- Moghimi, A., Rahmani, S., Zare, R., & Sadeghzadeh, M. (2014). Synthesis of 2-(2-fluorophenyl)-2-methylamino-cyclohexanone as a new ketamine derivative. Synthetic Communications, 44(14), 2021-2028.
- Morris, H., & Wallach, J. (2014). From PCP to MXE: a comprehensive review of the non‐medical use of dissociative drugs. Drug testing and analysis, 6(7-8), 614-632.
- Wallach, J. V. (2014). Structure activity relationship (SAR) studies of arylcycloalkylamines as N-methyl-D-aspartate receptor antagonists (Doctoral dissertation, University of the Sciences in Philadelphia).
- Colestock, T., Wallach, J., Mansi, M., Filemban, N., Morris, H., Elliott, S. P., … & Adejare, A. (2018). Syntheses, analytical and pharmacological characterizations of the ‘legal high’4-[1-(3-methoxyphenyl) cyclohexyl] morpholine (3-MeO-PCMo) and analogues. Drug Testing and Analysis, 10(2), 272-283.
- Giros, B., El Mestikawy, S., Godinot, Nathalie., Zheng, Keqin., Han, Hong., Yang-Feng, Teresa., & Caron, M. G. (1992). Cloning, pharmacological characterization, and chromosome assignment of the human dopamine transporter. Molecular pharmacology, 42(3), 383-390.
- Wallach, J., & Brandt, S. D. (2018). Phencyclidine-based new psychoactive substances. New Psychoactive Substances, 261-303.
- Kamenka, J. M., Chiche, B., Goudal, R., Geneste, P., Vignon, J., Vincent, J. P., & Lazdunski, M. (1982). Chemical synthesis and molecular pharmacology of hydroxylated 1-(1-phenylcyclohexyl) piperidine derivatives. Journal of Medicinal Chemistry, 25(4), 431-435.
- Wallach, J., Kang, H., Colestock, T., Morris, H., Bortolotto, Z. A., Collingridge, G. L., … & Adejare, A. (2016). Pharmacological investigations of the dissociative ‘legal highs’ diphenidine, methoxphenidine and analogues. PLoS One, 11(6), e0157021.
- Roth, B. L., Gibbons, S., Arunotayanun, W., Huang, X. P., Setola, V., Treble, R., & Iversen, L. (2013). The ketamine analogue methoxetamine and 3-and 4-methoxy analogues of phencyclidine are high affinity and selective ligands for the glutamate NMDA receptor. PLoS One, 8(3), e59334.
- Smith, R. C., Meltzer, H. Y., Arora, R. C., & Davis, J. M. (1977). Effects of phencyclidine on [3H] catecholamine and [3H] serotonin uptake in synaptosomal preparations from rat brain. Biochemical pharmacology, 26(15), 1435-1439.
- Vaupel, D. B., McCoun, D. O. N. A. L. D., & Cone, E. J. (1984). Phencyclidine analogs and precursors: rotarod and lethal dose studies in the mouse. Journal of Pharmacology and Experimental Therapeutics, 230(1), 20-27.
- Maddox, V. H., Godefroi, E. F., & Parcell, R. F. (1965). The synthesis of phencyclidine and other 1-arylcyclohexylamines. Journal of medicinal chemistry, 8(2), 230-235.
- Geneste, P., JM, K., & SN, U. (1979). Détermination conformationnelle de dérivés de la phencyclidine en vue d’une corrélation structure-activité.
- Gole, D. J., Pirat, J. L., & Domino, E. (1987, September). Analysis of Phencyclidine (PCP and other Structurally Related Compounds by Gas Chromatography-Mass Spectronomy (GC-MS). Pharmacology Biochemistry and Behavior. (Vol. 28, No. 1, pp. 125-125). The Boulevard, Langford Lane, Kidlington, Oxford, England, OX51GB: Pergamon-Elsevier Science Ltd.
- Chaudieu, I., Vignon, J., Chicheportiche, M., Kamenka, J. M., Trouiller, G., & Chicheportiche, R. (1989). Role of the aromatic group in the inhibition of phencyclidine binding and dopamine uptake by PCP analogs. Pharmacology Biochemistry and Behavior, 32(3), 699-705.
- Linders, J., Furlano, D. C., Mattson, M. V., Jacobson, A. E., & Rice, K. C. (2010). Synthesis and preliminary biochemical evaluation of novel derivatives of PCP. Letters in Drug Design & Discovery, 7(2), 79-87.
- Robins, E. G., Zhao, Y., Khan, I., Wilson, A., Luthra, S. K., & Årstad, E. (2010). Synthesis and in vitro evaluation of 18F-labelled S-fluoroalkyl diarylguanidines: novel high-affinity NMDA receptor antagonists for imaging with PET. Bioorganic & Medicinal Chemistry Letters, 20(5), 1749-1751.
- Menzies, E. L., Hudson, S. C., Dargan, P. I., Parkin, M. C., Wood, D. M., & Kicman, A. T. (2014). Characterizing metabolites and potential metabolic pathways for the novel psychoactive substance methoxetamine. Drug testing and analysis, 6(6), 506-515.
- Lundberg, G. D., Gupta, R. C., & Montgomery, S. H. (1976). Phencyclidine. Clinical toxicology, 9(4), 503-511.
- McCarron, M. M., Schulze, B. W., Thompson, G. A., Conder, M. C., & Goetz, W. A. (1981). Acute phencyclidine intoxication: incidence of clinical findings in 1,000 cases. Annals of emergency medicine, 10(5), 237-242.
- Olney, J. W., Labruyere, J., & Price, M. T. (1989). Pathological changes induced in cerebrocortical neurons by phencyclidine and related drugs. Science, 244(4910), 1360-1362.
- Fujikawa, D. G. (1995). Neuroprotective effect of ketamine administered after status epilepticus onset. Epilepsia, 36(2), 186-195.
- Xiong, Z., Chang, L., Qu, Y., Pu, Y., Wang, S., Fujita, Y., … & Hashimoto, K. (2020). Neuronal brain injury after cerebral ischemic stroke is ameliorated after subsequent administration of (R)-ketamine, but not (S)-ketamine. Pharmacology Biochemistry and Behavior, 191, 172904.
- A A Michely, J., K Manier, S., T Caspar, A., D Brandt, S., Wallach, J., & H Maurer, H. (2017). New psychoactive substances 3-methoxyphencyclidine (3-MeO-PCP) and 3-methoxyrolicyclidine (3-MeO-PCPy): metabolic fate elucidated with rat urine and human liver preparations and their detectability in urine by GC-MS,“LC-(high resolution)-MSn” and “LC-(high resolution)-MS/MS”. Current Neuropharmacology, 15(5), 692-712.
- A A Michely, J., K Manier, S., T Caspar, A., D Brandt, S., Wallach, J., & H Maurer, H. (2017). New psychoactive substances 3-methoxyphencyclidine (3-MeO-PCP) and 3-methoxyrolicyclidine (3-MeO-PCPy): metabolic fate elucidated with rat urine and human liver preparations and their detectability in urine by GC-MS,“LC-(high resolution)-MSn” and “LC-(high resolution)-MS/MS”. Current Neuropharmacology, 15(5), 692-712.
- Dani, J. A. (2015). Neuronal nicotinic acetylcholine receptor structure and function and response to nicotine. International review of neurobiology, 124, 3-19.
- Ho, T. N., Abraham, N., & Lewis, R. J. (2020). Structure-function of neuronal nicotinic acetylcholine receptor inhibitors derived from natural toxins. Frontiers in Neuroscience, 14, 609005.
- Wang, H. Y., Lee, D. H., Davis, C. B., & Shank, R. P. (2000). Amyloid peptide Aβ1‐42 binds selectively and with picomolar affinity to α7 nicotinic acetylcholine receptors. Journal of neurochemistry, 75(3), 1155-1161.
- Freedman, R., Hall, M., Adler, L. E., & Leonard, S. (1995). Evidence in postmortem brain tissue for decreased numbers of hippocampal nicotinic receptors in schizophrenia. Biological psychiatry, 38(1), 22-33.
- D’Andrea, M. R., & Nagele, R. G. (2006). Targeting the alpha 7 nicotinic acetylcholine receptor to reduce amyloid accumulation in Alzheimer’s disease pyramidal neurons. Current pharmaceutical design, 12(6), 677-684.
- Quik, M., Bordia, T., Huang, L., & Perez, X. (2011). Targeting nicotinic receptors for Parkinson’s disease therapy. CNS & Neurological Disorders-Drug Targets (Formerly Current Drug Targets-CNS & Neurological Disorders), 10(6), 651-658.
- Philip, N. S., Carpenter, L. L., Tyrka, A. R., & Price, L. H. (2010). Nicotinic acetylcholine receptors and depression: a review of the preclinical and clinical literature. Psychopharmacology, 212(1), 1-12.
- Buisson, B., & Bertrand, D. (2002). Nicotine addiction: the possible role of functional upregulation. Trends in pharmacological sciences, 23(3), 130-136.
- Lewis, R. J., & Garcia, M. L. (2003). Therapeutic potential of venom peptides. Nature reviews drug discovery, 2(10), 790-802.
- Jennings, K. R., Brown, D. G., & Wright, D. P. (1986). Methyllycaconitine, a naturally occurring insecticide with a high affinity for the insect cholinergic receptor. Experientia, 42(6), 611-613.
- Wintersteiner, O., & Dutcher, J. D. (1943). Curare alkaloids from Chondodendron tomentosum. Science, 97(2525), 467-470.
- Matsubayashi, H., Alkondon, M., Pereira, E. F., Swanson, K. L., & Albuquerque, E. X. (1998). Strychnine: a potent competitive antagonist of α-bungarotoxin-sensitive nicotinic acetylcholine receptors in rat hippocampal neurons. Journal of pharmacology and experimental therapeutics, 284(3), 904-913.
- Utkin, Y. N. (2013). Three-finger toxins, a deadly weapon of elapid venom–milestones of discovery. Toxicon, 62, 50-55.
- Orhurhu, V. J., Vashisht, R., Claus, L. E., & Cohen, S. P. (2019). Ketamine toxicity.
- Jewett, B. E., & Thapa, B. (2020). Physiology, NMDA Receptor. In StatPearls [Internet]. StatPearls Publishing.
- McRoberts, J. A., Coutinho, S. V., Marvizón, J. C. G., Grady, E. F., Tognetto, M., Sengupta, J. N., … & Mayer, E. A. (2001). Role of peripheral N-methyl-D-aspartate (NMDA) receptors in visceral nociception in rats. Gastroenterology, 120(7), 1737-1748.
- Mota, S. I., Ferreira, I. L., & Rego, A. C. (2014). Dysfunctional synapse in Alzheimer’s disease–A focus on NMDA receptors. Neuropharmacology, 76, 16-26.
- Davies, S., & Ramsden, D. B. (2001). Huntington’s disease. Molecular Pathology, 54(6), 409.
- Zhou, Q., & Sheng, M. (2013). NMDA receptors in nervous system diseases. Neuropharmacology, 74, 69-75.
- Molero, P., Ramos-Quiroga, J. A., Martin-Santos, R., Calvo-Sánchez, E., Gutiérrez-Rojas, L., & Meana, J. J. (2018). Antidepressant efficacy and tolerability of ketamine and esketamine: a critical review. CNS drugs, 32(5), 411-420.
- Noreña, A. J. (2015). Revisiting the cochlear and central mechanisms of tinnitus and therapeutic approaches. Audiology and Neurotology, 20(Suppl. 1), 53-59.
- Valentino, R. J., & Volkow, N. D. (2018). Untangling the complexity of opioid receptor function. Neuropsychopharmacology, 43(13), 2514-2520.
- Carroll, F. I., Abraham, P., Parham, K., Bai, X., Zhang, X., Brine, G. A., … & Bowen, W. D. (1992). Enantiomeric N-substituted N-normetazocines: a comparative study of affinities at. sigma., PCP, and. mu. opioid receptors. Journal of medicinal chemistry, 35(15), 2812-2818.
- Williams, N. R., Heifets, B. D., Blasey, C., Sudheimer, K., Pannu, J., Pankow, H., … & Schatzberg, A. F. (2018). Attenuation of antidepressant effects of ketamine by opioid receptor antagonism. American Journal of Psychiatry, 175(12), 1205-1215.
- Contet, C., Kieffer, B. L., & Befort, K. (2004). Mu opioid receptor: a gateway to drug addiction. Current opinion in neurobiology, 14(3), 370-378.
- Pradhan, A. A., Befort, K., Nozaki, C., Gavériaux-Ruff, C., & Kieffer, B. L. (2011). The delta opioid receptor: an evolving target for the treatment of brain disorders. Trends in pharmacological sciences, 32(10), 581-590.
- Van’t Veer, A., & Carlezon, W. A. (2013). Role of kappa-opioid receptors in stress and anxiety-related behavior. Psychopharmacology, 229(3), 435-452.
- Sheffler, D. J., & Roth, B. L. (2003). Salvinorin A: the ‘magic mint’ hallucinogen finds a molecular target in the kappa opioid receptor. Trends in pharmacological sciences, 24(3), 107-109.
- Usiello, A., Baik, J. H., Rougé-Pont, F., Picetti, R., Dierich, A., LeMeur, M., … & Borrelli, E. (2000). Distinct functions of the two isoforms of dopamine D2 receptors. Nature, 408(6809), 199-203.
- Hall, H., Sedvall, G., Magnusson, O., Kopp, J., Halldin, C., & Farde, L. (1994). Distribution of D1-and D2-dopamine receptors, and dopamine and its metabolites in the human brain. Neuropsychopharmacology, 11(4), 245-256.
- Gruner, J. A., Marcy, V. R., Lin, Y. G., Bozyczko-Coyne, D., Marino, M. J., & Gasior, M. (2009). The roles of dopamine transport inhibition and dopamine release facilitation in wake enhancement and rebound hypersomnolence induced by dopaminergic agents. Sleep, 32(11), 1425-1438.
- Morilak, D. A., Barrera, G., Echevarria, D. J., Garcia, A. S., Hernandez, A., Ma, S., & Petre, C. O. (2005). Role of brain norepinephrine in the behavioral response to stress. Progress in Neuro-Psychopharmacology and Biological Psychiatry, 29(8), 1214-1224.
- Gabay, S., Pertzov, Y., & Henik, A. (2011). Orienting of attention, pupil size, and the norepinephrine system. Attention, Perception, & Psychophysics, 73(1), 123-129.
- Mandela, P., & Ordway, G. A. (2006). The norepinephrine transporter and its regulation. Journal of neurochemistry, 97(2), 310-333.
- Owens, M. J., & Nemeroff, C. B. (1994). Role of serotonin in the pathophysiology of depression: focus on the serotonin transporter. Clinical chemistry, 40(2), 288-295.
- De Felice, L. J. (2016). A current view of serotonin transporters. F1000Research, 5.
- Sørensen, A., Ruhé, H. G., & Munkholm, K. (2022). The relationship between dose and serotonin transporter occupancy of antidepressants—a systematic review. Molecular psychiatry, 27(1), 192-201.
- Maurice, T., & Su, T. P. (2009). The pharmacology of sigma-1 receptors. Pharmacology & therapeutics, 124(2), 195-206.